Epileptic Disorders
MENUA triad of infantile spasms, nystagmus and a focal tonic seizure Volume 20, issue 4, August 2018

Epileptic spasms (ES) affect one in 2,000 infants and occur in a number of epilepsy syndromes, with West syndrome being the most frequently described (Kossoff, 2010). As outlined in the International League Against Epilepsy (ILAE) 2017 classification, ES can be focal or generalized, depending on both clinical and electrographic correlates, or of unknown onset in the absence of distinguishing features (Fisher et al., 2017). Although the clinical semiology of ES has been described at length, typically consisting of a sudden flexion, extension or mixed flexion-extension of proximal and truncal muscles, subtle forms may occur, particularly in infants, making clinical classification somewhat challenging. Furthermore, combined seizure types occurring sequentially can equally hamper the clinician's task of classifying seizures. These clinical challenges can have consequences on medical and surgical management in such cases.
ES occurring in sequence with other seizure types has been previously described, including generalized atonic seizures (Xue et al., 2017) and other focal motor seizures (Lee et al., 2015). ES associated with focal motor seizures, previously referred to as “focal spasms” (Caraballo et al., 2003), has been the topic of various reports in an attempt to identify the cortical trigger and underlying epileptogenic networks at the forefront of seizure genesis. So far, these mechanisms have not been fully elucidated.
Ictal nystagmus, originating from both the saccade and slow pursuit areas, has been described and is more frequently associated with seizures arising from posterior cortical areas (Lee et al., 2014). To our knowledge, the sequential combination of epileptic spasms, nystagmus, and a focal motor seizure has not been previously described. We report the case of an infant boy who presented all three seizure types in clusters and provide video illustration of a typical event. The clinical and electrographic components in this case reveal the complexity of seizure classification at times, and perhaps help to outline possible underlying brain structures involved in the different seizure types.
Case study
This five-month-old boy was born at term by vaginal delivery without any gestational or perinatal complications. He initially presented with normal psychomotor development, smiling within the first month, and rolling from the age of four months. His parents had, however, noticed poor visual pursuit. No relevant findings were identified from the family history.
At three months, the patient started presenting paroxysmal episodes, typically upon awakening, with each episode consisting of loss of contact, eye deviation to the left, brief bilateral symmetric flexor contraction of the neck and upper extremities, with subsequent onset of a vertical nystagmus, and, following the more sustained component of the symmetric spasm, a focal tonic contraction of the right upper arm lasting over ten seconds (video sequence). These episodes occurred in clusters lasting for a few minutes, with three to four clusters per day noted initially. His behaviour was reported as normal between the events.
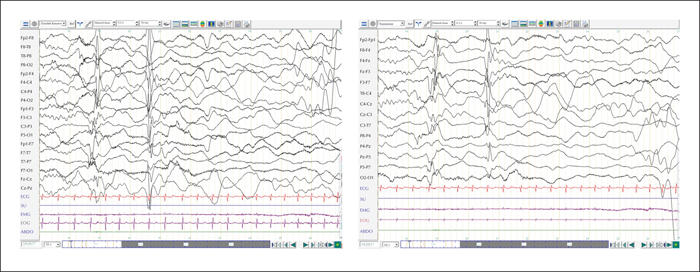
Upon first referral, his weight, height, and head circumference were normal and his neurological examination showed no focal signs. Ophthalmological examination showed unsustained visual fixation, without spontaneous nystagmus. The first EEG recording prior to treatment showed a background activity consisting of high-voltage delta and theta rhythms (3-4 Hz), predominantly in the right hemisphere (figure 1A) with multifocal interictal sharp waves (at frontal F4, temporal T8, and parieto-occipital P8 electrodes). These sharp waves appeared independent, isolated, and polymorphic, and were accentuated during sleep. Frontal sleep spindles were seen bilaterally and asynchronously during sleep. Seizures were recorded and the ictal EEG showed a characteristic diffuse electrodecrement, preceded by a frontally dominant slow wave (figure 1B), more pronounced on the right side. This was followed by bilateral rhythmic fast activity of low amplitude, predominant in the fronto-central regions (figure 1C), that merged into a rhythmic polyspike-and-wave pattern with an electrodecrement (figure 1D). The temporal relationship between clinical signs and EEG findings is summarized in table 1. An extensive biological blood and urine work-up, including electrolytes, kidney and liver function, as well as metabolic profiles, was normal. No focal or global cortical or subcortical lesion was identified on 1.5T brain MRI. Genetic investigations included a normal array CGH, and next-generation sequencing with bioinformatic analysis for targeted epilepsy and developmental delay genes was negative.
We concluded a diagnosis of West syndrome with focal ES and a treatment of vigabatrin was initiated. Within the next 48 hours, all seizures ceased. One week after treatment initiation, the EEG already showed significant improvement. The background had normalized with a symmetric posterior theta activity and postero-anterior gradient. During sleep, there were many synchronous and asynchronous sleep spindles. Rare epileptiform spike-and-wave complexes were identified over the right temporal region. EEG had fully normalized four months after onset. At seven months after onset, the infant had a normal head circumference (44.5 cm; P: 25-50) and normal muscle tone and motricity; he was able to sit and demonstrated numerous vocal sounds. Although progress was noted on eye examination, the gaze was still not sustained for more than a few seconds, and a discrete alternating divergent strabismus was noted. Vigabatrin could be withdrawn over a two month-period without seizure recurrence. At the last follow-up visit, aged 14 months, he continued to show improvement in his gross and fine motor skills with mild developmental delay and discrete asymmetry in favour of his right side. Intermittent alternating exotropia persisted as well as mild-to-moderate delay in visual motor integration skills.
Discussion
To the best of our knowledge, this is the first report of a triad of ictal symptoms associating ES, binocular nystagmus, and focal tonic seizures.
There is a need to further investigate the clinical expression of peculiar forms of ES in order to better ascertain their pathophysiology and mode of onset (Berg et al., 2010). Despite clearly outlined systems for classification of ES, clinical experience often leads to encounters with atypical characteristics that challenge the concepts laid down. ES can often be characterized as a focal seizure in the presence of cortical malformations identified on brain MRI (Watanabe et al., 2007; De Marchi et al., 2015), with tuberous sclerosis complex (TSC) being a common example. When spasm semiology appears bilateral and symmetric, associated focal seizures arising simultaneously or in sequence can raise suspicion of a localized onset. Such focality can be confirmed in hindsight upon seizure-free outcomes following cortical resection (Caraballo et al., 2016). Focality in our case was suggested at onset with left eye deviation and right frontal predominance of discharges on EEG, despite the fact that spasm contractions remained symmetric throughout the ictal event. The co-occurrence of several seizure types in a single ictal event provides a glimpse into the complex nature of the ongoing epileptogenic network at both cortical and subcortical levels. This co-occurrence of seizures appears more frequently in young infants (Pachatz et al., 2003), suggesting that it likely undergoes a maturational process.
Various explanations have been put forward to suggest a cortical or subcortical origin for ES although most studies agree on the likelihood of a cortical-subcortical circuitry, whereby a defective interaction leads to the facilitation or inducement of epileptiform activity (Lado and Moshe, 2002; Vigevano et al., 2001). This abnormal interaction, as the mainstay of ES, that would be somewhat dependent on the maturation of white matter connections, might explain the limited time frame during which spasms occur, particularly those in close temporal relationship with other focal seizures during infancy. These cases of seizure co-occurrence might assist in pinpointing the origin of ES with additional semiology providing better localization.
Previous reports suggest that seizures associated with ictal nystagmus are often related to underlying lesions in the posterior part of the brain (Lee et al., 2014). Epileptiform discharges have been recorded in saccadic, optokinetic or pursuit cortical regions, but whether such discharges were propagated from deeper brainstem structures remains difficult to assess with scalp EEG. Reported underlying aetiologies for ictal nystagmus vary and include focal structural disruptions such as focal cortical dysplasia or vascular disease, as well as systemic conditions, including hypoglycaemic encephalopathy, uremic encephalopathy, and mitochondrial disease. No such aetiology was identified in our patient.
Rhythmic vertical eye movements have also been described following myoclonic-tonic seizures in late-infantile epileptic encephalopathy (LIEE) with late-onset epileptic spasms and cryptogenic late-onset epileptic spasms (Nordli et al., 2006). The presence of vertical ictal nystagmus, as in our case, hints at possible subcortical involvement, specifically the brainstem, a structure previously hypothesized to be involved in the genesis of ES (Lado and Moshe, 2002; Avanzini et al., 2012). However, the abnormal visual pursuit that persisted after cessation of ES in this case might suggest involvement of a cortical visual network including the middle temporal, intraparietal, and frontal areas; a part of the cortico-ponto-cerebellum pathway (Thier and Ilg, 2005). The predominance of frontal discharges in the ictal recording on EEG and the co-occurrence of a focal motor seizure during the ictal cluster would suggest that the visual manifestations arose in the frontal region, such as the frontal eye field (FEF), as this participates in all types of eye movements, such as smooth pursuit and nystagmus. The pursuit area is situated deeper within the precentral sulcus and one recent study has shown that stimulation of the right FEF can instigate upward eye movement bilaterally (Kaiboriboon et al., 2012). The presence of sharp waves in the right temporal and parietal regions during the interictal state in our patient, as well as the delayed occurrence of the vertical nystagmus with regards to the frontal discharges and spasm onset, could suggest a temporo-parietal origin, although this is not evident on the ictal EEG recording and we found no previous citations of vertical nystagmus associated with temporo-parietal lesions in the literature.
The elaborate ictal semiology encountered in our case highlights the difficulty in seizure classification, at times, in the clinical setting. The combination of ES, nystagmus, and a focal tonic seizure during a single ictal event illustrates the complexity of the underlying epileptogenic network. Focal findings at onset with left eye deviation and prominent right frontal discharges suggest a lateralized frontal origin, and findings in the literature on visual ictal manifestations arising in the frontal eye region, including pursuit and nystagmus, would concur.
Acknowledgements and disclosures
We would like to thank the parents for enabling us to publish clinical information and video-recordings of their child, in order to advance our knowledge on epileptic spasms.
None of the authors have any conflict of interest to declare.