Epileptic Disorders
MENUA comprehensive clinico-pathological and genetic evaluation of bottom-of-sulcus focal cortical dysplasia in patients with difficult-to-localize focal epilepsy Volume 21, numéro 1, February 2019

Focal cortical dysplasia (FCD) are common histopathological lesions in children and adults with drug-resistant focal epilepsy (Blümcke et al., 2017), and hitherto classified into separate clinico-pathological subtypes (Blümcke et al., 2011). However, the aetiology and pathogenesis of most of these subtypes remain to be clarified (Najm et al., 2018). Such knowledge will be mandatory to also understand their variable occurrence in size, cellular phenotypes, brain localization and clinical presentation (Krsek et al., 2008; Lerner et al., 2009; Blümcke et al., 2010; Chassoux et al., 2012; Harvey et al., 2015). Continuous improvement in magnetic field strength for MRI diagnosis and the application of advanced post-processing analyses has significantly enhanced clinical identification of FCD subtypes in vivo, in particular, of FCD ILAE type II (Urbach et al., 2002; Besson et al., 2008; Bernasconi et al., 2011; Wagner et al., 2011; Mellerio et al., 2014; Wang et al., 2014). As a pertinent example, hyperintense MRI signalling from the lateral ventricle towards the crown of the gyrus was described as a “transmantle sign” (Barkovich et al., 1997), and mostly confirmed in FCD IIb and in the frontal lobe (Colombo et al., 2009; Colombo et al., 2012). Not all FCD II present, however, with a transmantle sign suggesting a larger clinico-pathological spectrum disorder or even separate disease entities. Two previous reports focused on FCD II located at the bottom of sulcus and highlighted these challenges in neuroimaging, clinical, and electroclinical presentation (Chassoux et al., 2012; Harvey et al., 2015).Tailored surgical resection was particular favourable, with 87-94% of reported patients (n=57) being completely seizure-free. Intriguingly, about 25% of patients did not reveal abnormal signals at initial MRI examination, and the combination of PET with MRI increased the detection rate (Chassoux et al., 2010).
With few exceptions, current research has failed to establish pathology-specific molecular biomarkers that clearly distinguish FCD subtypes (Guerrini et al., 2015). In the absence of adequate animal models, surgical brain tissue samples open the unique opportunity to further study tissue-specific signatures. A milestone in FCD research represented the identification of brain somatic mutations, germline mutations, or second-hit mosaic mutations activating the mTOR pathway in surgical brain specimens with histopathology-proven FCD type II (Jamuar et al., 2014; Scheffer et al., 2014; Baulac et al., 2015; D’Gama et al., 2015; Lim et al., 2015; Mirzaa et al., 2016; Moller et al., 2016; D’Gama et al., 2017; Ribierre et al., 2018). With only a third of published cases showing a genetic lesion (Marsan and Baulac, 2018), however, continuous efforts are required to improve our understanding of clinically-meaningful FCD subtypes and successful treatment strategies in the near future. The integration of clinical phenotypes with histopathology and genetic analysis is a powerful option, as recently proposed and already implemented by the WHO for the diagnosis of malignant gliomas and embryonal brain tumours (Louis et al., 2016).
Methods
Selection of patients
To investigate patients with difficult-to-localize epilepsy due to cortical dysplasia at the bottom of sulcus, we retrospectively reviewed the Cleveland Clinic Epilepsy Center's surgery database with patients who underwent invasive intracranial studies from 2004 to 2014 (as approved by the Cleveland Clinic Institutional Review Board). Inclusion of patients was based on the following criteria:
- –drug-resistant focal epilepsy;
- –a single MRI lesion restricted to the bottom of a sulcus;
- –no previous epilepsy surgery;
- –intracranial video-EEG monitoring prior to surgery;
- –no concomitant other diagnosis, such as tuberous sclerosis or brain tumour;
- –post-operative MRI available to assess the extent of the resection;
- –and histopathology slides available for post hoc microscopic review.
Magnetic resonance imaging (MRI)
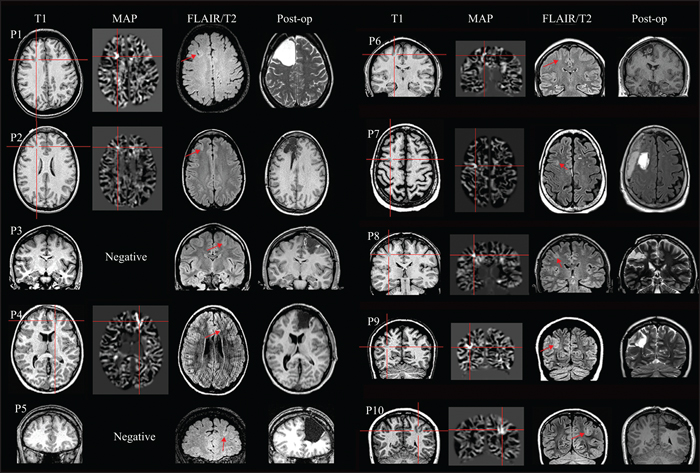
Four patients were imaged with a 3 T Siemens Trio/Skyra scanner (Erlangen, Germany) and six patients with a 1.5 T Siemens Avanto scanner (Erlangen, Germany). Sequence parameters at 3 T were: repetition time = 1,860 milliseconds, echo time = 3.4 milliseconds, inversion time = 1,100 milliseconds, flip angle = 10 degrees, band width = 130 kHz, slice thickness = 0.94 mm, no gap, and a 256 × 256 matrix providing isotropic voxels of 0.94 mm. Sequence parameters at 1.5 T were: repetition time = 11 milliseconds, echo time = 4.6 milliseconds, no inversion, flip angle = 20 degrees, band width = 130 kHz, slice thickness = 1.25 mm, no gap, and a 256 × 256 matrix providing 0.9 mm in-plane resolution. All MR images were reviewed by experienced board-certified neuroradiologists specialized in epileptology. Morphometric MRI analysis was available in one patient at the time of surgical evaluation and was retrospectively processed in the remaining nine patients. A voxel-based morphometric analysis program (MAP) was carried out in SPM (Wellcome Department of Cognitive Neurology, London, UK) and Matlab (MathWorks, Natick, Massachusetts) following established protocols (Huppertz et al., 2005). MAP was performed on T1-weighted MPRAGE sequence, and the grey-white junction output was examined in each patient with a z-score threshold of 4; the choice of threshold was consistent with previous reports (Wang et al., 2014, 2015) (figure 1). All 10 patients had FDG-PET. Ictal SPECT was successfully accomplished in six patients and subtraction ictal SPECT coregistered with MRI (SISCOM) was performed.
Neurophysiology
All patients had continuous scalp video-EEG monitoring to confirm the focal epilepsy and characterize the seizure semiology (table 1). Following the initial non-invasive evaluation, a recommendation for an invasive intracranial video-EEG evaluation was made during the Cleveland Clinic Epilepsy Center multidisciplinary patient management conference in all 10 patients for three reasons:
- –ictal EEG findings and FDG-PET or ictal SPECT was discordant in five patients (table 1);
- –eloquent cortical areas had to be mapped for the definition of surgical resection borders in four patients;
- –and BOS-FCD was not unambiguously accepted by the group in one patient (Patient 5) (table 1).
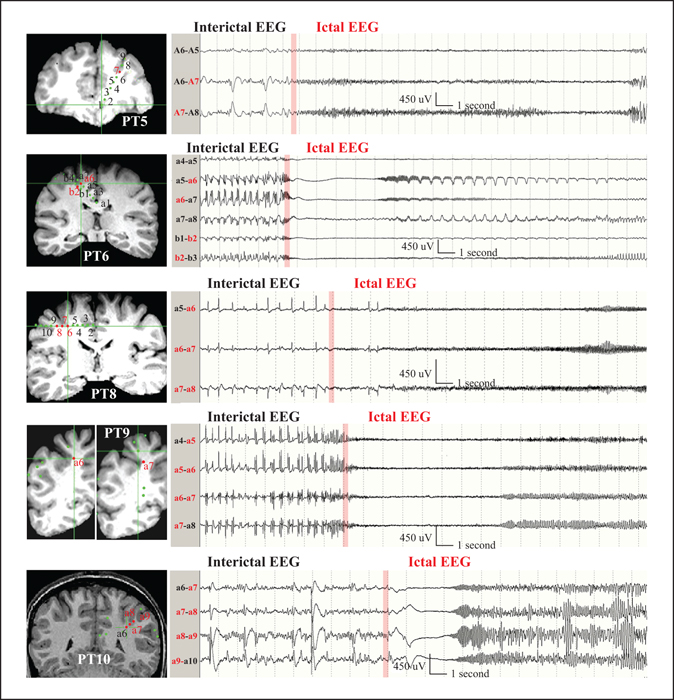
Eight patients had an implantation of subdural grid electrodes (SDG) together with intracerebral depth electrodes targeting the BOS-FCD of interest. The placement of depth electrodes and their 3D spatial correlation with the MRI-identified lesion was verified through co-registration of post-implantation volume acquisition CT scans and preimplantation high-resolution MRI volume acquisition sequences (figure 2). One patient had SDGs without depth electrode implantation (Patient 3) (table 1). Patient 8 had stereotactic implantation of depth electrodes according to the SEEG methodology, as previously described (Gonzalez-Martinez et al., 2014).
Neurosurgery
Surgical resection strategies were discussed following the invasive evaluation at our patient management conference, integrating all available data from MRI analysis and neurophysiological recordings. Post hoc analysis of the extent of surgical resection was obtained from post-surgical MRI. Post-operative seizure outcome was assessed during regular outpatient visits using Engel's classification scale (Engel et al., 1993).
Immunohistochemistry
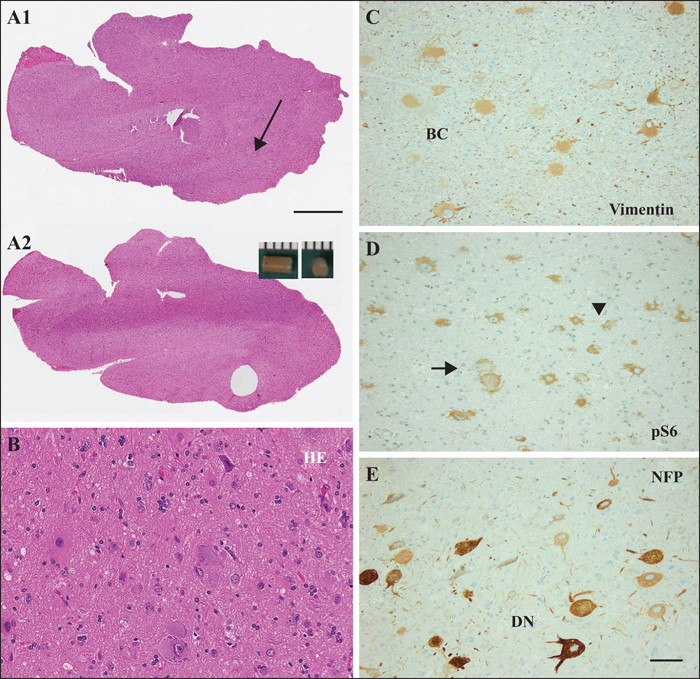
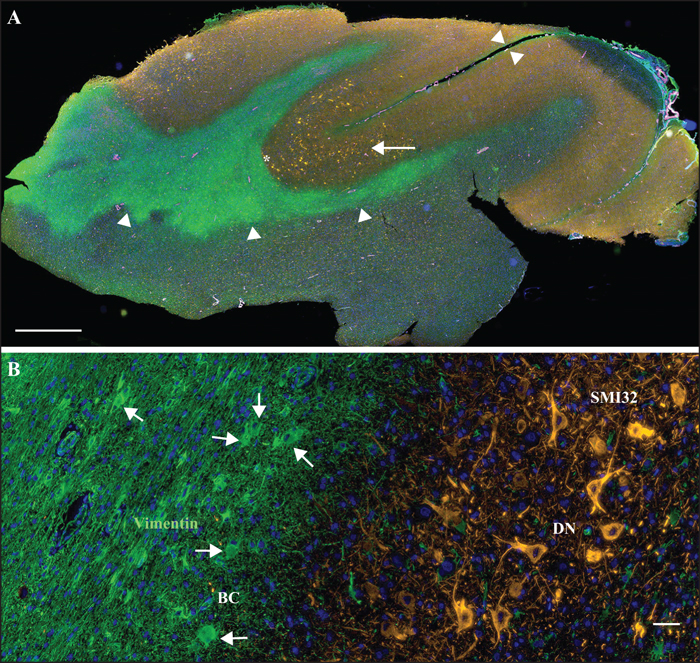
For histopathological diagnosis, surgical specimens were formalin-fixed and paraffin-embedded. Post hoc review of all surgical tissue was based on immunohistochemical stainings to visualize architectural dysplasia (Blümcke et al., 2016) using antibodies directed against NeuN (clone A60, Chemicon, USA) and MAP2 (clone HM2, DAKO, Denmark), to visualize dysmorphic neurons with antibodies directed against non-phosphorylated neurofilaments (clone SMI32, Covance, USA) or balloon cells with antibodies directed against vimentin (polyclonal antibody V9, DAKO, Denmark) (see also Blümcke et al. [2016]). Immunohistochemical detection of the phospho-S6 epitope (clone ser235/236, Cell Signaling Technology, USA) was used to demonstrate an activated mTOR pathway. Areas with highest content of abnormal cells were identified on the H&E section, and the same area labelled in the FFPE bloc. In nine patients, FFPE tissue was micro-dissected using a 2-mm diameter large punching needle (figure 3). DNA extraction from the FFPE tissue punch was performed using customized protocols for small FFPE tissue (Qiagen, Germany) with sufficient DNA available for deep sequencing in five patients. Semi-quantitative cell measurements were performed as following: a HE stained section was prepared before and after the tissue punch and fully digitized using whole slide digital imaging (3DHistech, Hungary). All cells within 1 mm2 of the punched area were counted from the computer screen (range: 171-673 cells). FCD-specific cells referred to dysmorphic neurons in FCD IIa and IIb and balloon cells in FCD IIb and were counted from the same region of interest (range: 18-80 cells). Results were expressed as percentage of FCD-specific from total cells.
Genetic analysis
To perform the targeted sequencing, we used the Agilent SureSelect Custom Enrichment Kit for library preparation of 166 self-selected genes. Library preparation was conducted according to the manufacturer's protocols and subsequent paired-end library sequencing was performed using the Illumina HiSeq4000. The targeted genes included those encoding proteins of the mTOR and PI3K-AKT signalling pathway, genes associated with low-grade brain tumours, and genes associated with epilepsy. The list of mTOR pathway genes was derived from the Kegg Pathway (ID: hsa04150). The PI3K-AKT pathway genes were derived from RT2 Profiler PCR Array (Qiagen, Product no.: 330231). Genes associated with low-grade brain tumours were derived from the recent literature and epilepsy genes were derived from the EpiPM Consortium review in 2015 (Vogelstein et al., 2013; EpiPMConsortium, 2015). The full list of included genes is disclosed in supplementary table 1.
For bioinformatic analysis, we generated analysis-ready bam files using BWA to map reads to the human genome reference build GRCh37 (Li and Durbin, 2009). GATK was used to mark duplicated reads (McKenna et al., 2010), perform local realignment, recalibrate the base quality scores, and call SNPs and short indels together with SAM tools and Dindel (Li et al., 2009; Albers et al., 2011). In addition, we used Platypus to call low allele frequency variants (Rimmer et al., 2014). We used the human reference genome build GRCh37 and annotated variant functional consequences and population allele frequencies using wANNOVAR (excessed: 12/2016; http://wannovar.wglab.org). We removed non-protein-coding variants and variants present in individuals from the general population with allele frequency >0.1% to enrich for rare variants of large effect. Variants passing our applied filters, were manually inspected for sequencing and variant calling quality using the Integrative Genomics Viewer (Robinson et al., 2011). The manual evaluation was conducted by three independent scientists. Variant pathogenicity was assessed in accordance with 28 criteria defined by guidelines of the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). We used the online tool, InterVar, to facilitate the variant interpretation process (Li and Wang, 2017).
Results
Patient data
Ten patients fulfilled our inclusion criteria; four males and six females. None of the patients had any neurological deficit at clinical examination. One patient had mild developmental delay (Patient 4) (table 1). All patients presented with stereotyped seizures (see table 1), without a history of status epilepticus, infantile spasms or febrile seizures. Age at epilepsy onset ranged from 0.75 to 14 years (mean: 7.2 years). Age at time of surgery ranged from 13 to 59 years (mean: 33.2 years). Epilepsy duration was between one and 46 years (mean: 23.4 years). None of the patients had a history of pre- or perinatal injuries except for one patient who was born two months premature (Patient 7) (table 1). Clinical histories and seizure description of all patients included in the study are summarized in table 1.
Imaging data
BOS-FCD was identified in all patients by epilepsy expert neuroradiology review (figure 1). Six lesions were located in the superior frontal sulcus, one lesion in the inferior frontal sulcus, one lesion in the central sulcus, and two lesions in the intraparietal sulcus. Focal cortical thickening and blurring of the grey-white matter junction at the bottom of a sulcus was a common MRI finding in all patients, as illustrated in figure 1. These abnormalities were best visible on FLAIR sequences. Eight out of 10 patients had concordant signal changes on T1w images (figure 1). Blurring of the grey-white matter junction was evident on T1w images in four patients by visual inspection (Patients 1, 4, 9, and 10) (table 1), and eight patients showed positive MAP foci. Blurring of the grey-white matter junction on T1w images became evident in four patients only after MAP analysis with a z-score set >4 (Patients 2, 6, 7, and 8). No T1w signal changes were observed visually or by MAP analysis in the remaining two patients (Patients 3 and 5). A funnel-shaped, subcortical hyper-intensity tapering abnormality towards the ventricular surface (transmantle sign) was seen in three patients (Patients 1, 8 and 9) (see figure 1).
Scalp video-EEG monitoring
Habitual seizures were documented in all patients by scalp video-EEG-monitoring. Semiology could not be, however, correlated simply with lobar location of the BOS-FCD (table 1). As an example, most of the auras were non-specific. Secondly, presence of auras with eye involvement could be anatomically misleading in both patients (Patients 9 and 10) with intraparietal BOS-FCD. Ictal EEG patterns were non-localizable in two patients (Patients 1 and 2) (table 1), regional lobar for two patients in one brain region (Patients 4 and 8) and for two patients with two adjacent brain regions (Patients 9 and 10), and at the midline vertex region in two patients (Patients 3 and 6). Interictal sharp waves were absent in five patients and did not add localizing value in non-localizing ictal EEGs.
FDG-PET and ictal SPECT
Interictal FDG-PET studies were performed in all patients (table 1); only five out of 10 patients exhibited focal hypometabolism concordant with the lesion detected by MRI, whereas the remaining five patients had non-contributory results, including two patients with discordant PET localization. In four out of six patients with successful ictal SPECT and SISCOM, the area of hyperperfusion was concordant with the MRI-visible BOS-FCD (table 1).
Invasive video-EEG monitoring
At the time of surgical evaluation, all 10 patients proceeded to invasive EEG evaluation based on the decision of the patient management conference at that time (table 1): non-localizing ictal EEG in three patients, atypical semiology associated with the lesion in one patient, MRI lesion was not unanimously convinced by the committee in one patient, discordant PET hypometabolism in one patient, and functional mapping was recommended in four patients.
Habitual seizures were documented in all patients also by invasive video-EEG monitoring. Ictal EEG onset was mapped to depth electrode contacts localized in the BOS-FCD or in areas sampled adjacent to the lesion in eight patients (figure 2). In the remaining two patients (Patient 2 with no depth electrode contacts in the proximity of the lesion or depth of sulcus, and Patient 3 with SDG electrodes only), ictal EEG onset was recorded from electrodes placed in the gyral crown of the sulci harbouring the lesion.
The depth electrodes were located within or in close vicinity to the BOS-FCD in seven patients (table 1). Only in Patient 2, co-registration revealed the depth electrode not inside or close to the lesion. Histopathology analysis confirmed the iEEG trajectory in close vicinity to the FCD in one patient (data not shown). Intracranial EEG recordings with depth electrode contacts in bottom-of-sulcus lesions showed continuous interictal discharge patterns consisting of rhythmic (1-3-Hz) spikes/polyspikes and waves. The rhythmic spiking pattern was intermixed with low-amplitude fast discharges (figure 2). Extraoperative video and EEG recordings captured the patients’ typical clinical seizures with stereotyped ictal EEG onset patterns that were localized at the depth electrode contacts in or near the BOS-FCD (figure 2, also in reference to figure 1). All EEG seizures transitioned from continuous pre-ictal rhythmic epileptiform discharges to tonic fast frequency discharges of variable voltage for durations ranging from 15 to 25 seconds (figure 2).
Surgical resection and seizure outcome
Neurosurgical resection of the lesion was limited to overlying and surrounding cortex (i.e. lesionectomy) in seven patients and corticoectomy with resection of the lesion plus adjacent gyri was performed in three patients (table 1). Figure 1 illustrates the anatomy of the resection in all patients (post-operative MRI). Eight patients became free of seizures and auras (Engel Class 1A) (table 1) with a mean follow-up time of six years (2.5 years to 11 years). One patient (Patient 7) did not return for follow-up. One patient (Patient 10) had incomplete resection due to the epileptic zone overlapping with eloquent cortex, as determined by extraoperative brain stimulation via subdural grids and depth electrodes, and this patient continued to have seizures (Engel Class III).
Histopathological findings
All surgical specimens were histopathologically and immunohistochemically reviewed and classified as FCD ILAE type II, with a combination of dysmorphic neurons and balloon cells (FCD IIb) in six patients and dysmorphic neurons only (FCD IIa) in three patients (table 1). Only small tissue fragments were submitted for pathological examination in one patient and microscopic review remained inconclusive (Patient 8). The gross neuroanatomical presentation of FCD IIb can be demonstrated best using immunohistochemical labelling of surgically well-preserved specimens with the characteristic presentation of vimentin-immunoreactive balloon cells and neurofilament-accumulating dysmorphic neurons (figures 3, 4). In addition, immunohistochemical labelling of the phospho-S6 epitope revealed specific staining in all specimens.
Genetic findings
Targeted next-generation sequencing (NGS) achieved a mean coverage of 245 × (SD = 60) across the target genes, with 96.3% (SD = 0.95) of bases covered at 50 × (supplementary table 1). The microdissected patient samples harboured a mean fraction of dysplastic cells of 8.6% (SD=1.4). Given our sequencing coverage and the assumption that all dysplastic cells should carry the variant, we would have been able to detect 99% of all brain somatic variants with Platypus in these cells (Richards et al., 2015). We screened for coding variants in 166 candidate genes (supplementary table 1) and did not identify brain somatic variants. DNA obtained from blood leucocytes was, however, not available in this retrospective analysis to confirm germline origin. The variants were identified in eight genes and comprised nine exonic heterozygous missense variants (mean allele frequency = 47%; SD = 3.6) and one frameshift insertion introducing a stop codon. All nine missense variants were classified as ‘variants of uncertain significance’ (VUS) using state-of-the-art guidelines in the field (Richards et al., 2015). The frameshift insertion (NM_001136029, p.Asp1075Glufs*3) introduces a premature stop codon in DEPDC5, likely leading to haploinsufficiency and was classified as ‘likely pathogenic’ for the epilepsy according to recommended ACMG guidelines and current epilepsy literature (Ishida et al., 2013; Lal et al., 2014; Epi4Kconsortium, 2017).
Discussion
Our comprehensive analysis of 10 patients with difficult-to-localize BOS-FCD confirms previous studies with a “syndromic description” of FCD ILAE type II:
- –seizure onset at preschool or school age;
- –mostly of frontal localization;
- –stereotyped seizures;
- –distinct MRI features;
- –intrinsic epileptogenicity;
- –favourable postsurgical seizure outcome following complete resection of the epileptic region;
- –and exclusivity of FCD type II with immunohistochemical or genetic evidence for activation of the mTOR pathway.
The fact that FCD-BOS lesions are small and localized to the bottom of sulcus suggested, however, a later occurrence of a (presumably) genetically acquired pathogen during the estimated 32 mitotic cycles of cortical brain development, compared to FCD II lesions involving a larger cortical area or extending even to hemimegalencephaly (D’Gama et al., 2017; Blümcke and Sarnat, 2016).
NGS analysis of a panel of 166 mTor, PIK3/Akt, and other epilepsy-related genes detected a likely pathogenic, epilepsy-associated variant in DEPDC5 (terminology used according to ACMG guidelines) in only one out of five patients studied. This is consistent with previous studies, which reported ‘likely pathogenic’ variants in mTOR pathway-associated genes in only 25% (SD=40) of patients with histopathologically confirmed FCD II (Marsan and Baulac, 2018). The majority of variants have been reported as brain somatic with allele frequencies of 1-12.6%, and predominantly affecting the MTOR gene (Baulac et al., 2015; D’Gama et al., 2015; Lim et al., 2015; Mirzaa et al., 2016; Moller et al., 2016; D’Gama et al., 2017). One study reported germline DEPDC5 mutations in cases of BOS-FCD, which further stressed the association with mTORopathies (Scheffer et al., 2014). Recently described second-hit mosaic mutations may be another etiologic pathomechanism to be taken into consideration (Ribierre et al., 2018). In our present study, we detected a new pathogenic DEPDC5 heterozygous variant in one patient with FCD IIa. Stop codon-inducing germline variants in DEPDC5 have recently been shown to be present in 3% of patients (cohort: n=1187) with familial non-acquired focal epilepsy without cortical structural abnormalities and only in 0.05% of controls (cohort: n=3877; p=9.6 × 10−12) (Epi4Kconsortium, 2017). In addition, one study identified a somatic mutation in DEPDC5 in addition to an existing germline mutation (Baulac et al., 2015). The DEPDC5 variant identified in this study had an allele frequency of 41%. However, only 8.6% (SD=1.4) of cells shared a dysplastic phenotype by microscopic review, indicating that the identified p.Asp1075Glufs*3 DEPDC5 variant is unlikely to be only present in dysplastic cells. Unfortunately, we were not able to validate this prediction because blood samples were not available retrospectively. Further studies should clarify whether this DEPDC5 variant is causal for the epilepsy or if the epilepsy is secondary to FCDII. In all other BOS-FCD patients, no likely pathogenic or pathogenic variant was identified. However, our NGS coverage was, for the majority of samples, higher compared to previous reports (median: 243.72x vs. 180x), which should enable us to detect 99% of all somatic variants with variant allele frequencies of > 8%. Our results call for extended molecular/genetic investigations integrating ultra-deep exome-wide DNA and single-cell RNA sequencing, as well as methylome and proteomic analysis to identify a possible pathogenic cause(s). Future progress in precision medicine will build on such analysis to develop a targeted drug treatment for specific mTOR signalling molecules, in particular, when epilepsy surgery is not an option for a given patient.
Despite the fact that our study addressed only a small number of patients and any conclusion would need confirmation by larger and prospectively collected patient series, the comprehensive approach integrating genotype with phenotype analysis will help to consolidate the recognition of FCD-BOS in focal and difficult-to-localize epilepsies. Re-review of MRI and application of post-processing methodologies led to the identification of cortical dysplasia at BOS localization in all our patients, most often in the frontal or parietal lobes. Favourable outcome after neurosurgical resection, histopathological diagnosis of FCD II, and genetic testing helped to validate the clinical hypothesis. No other diagnostic modality added significant value in clinical management, as seen from a retrospective angle. In the future, MRI fingerprinting is the resolution for this population of patients (Ma et al., 2013).
Supplementary data
Supplementary table is available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
The work was supported by the European Union (FP7 DESIRE GA # 602531). We are thankful to Emily Kiefer (Cleveland) and Birte Rings (Erlangen) for their expert technical assistance.
None of the authors have any conflict of interest to declare.