Médecine thérapeutique / Pédiatrie
MENULes leucémies aiguës lymphoblastiques de l’enfant Volume 22, numéro 2, Avril-Mai-Juin 2019


Introduction
Les leucémies aiguësreprésentent environ 30 % de l’ensemble des cancers pédiatriques et sont ainsi la première cause de cancer chez l’enfant de moins de 15 ans. Au sein des leucémies aiguës, les formes lymphoblastiques sont largement prédominantes chez l’enfant puisqu’elles comptent pour près de 80 % de l’ensemble des leucémies aiguës. Les leucémies aiguës lymphoblastiques (LAL) se divisent en deux groupes selon une classification reposant sur l’expression de marqueurs membranaires, témoins de l’engagement des progéniteurs lymphoïdes dans les lignées B ou T.
Depuis sa première description en 1847 par le Dr Virshow qui donna son nom à cette pathologie (du grec leukós, blanc et haîma, sang) et les premiers essais thérapeutiques initiés aux États-Unis par Sydney Farber à la fin des années 40, les LAL représentent un modèle unique de réussite thérapeutique dans le champ de l’oncologie. Initialement associées à un pronostic irrémédiablement sombre, les LAL ont en effet vu leur prise en charge s’améliorer à partir des années 60 pour atteindre actuellement des survies globales proches de 90 %. Plusieurs facteurs ont contribué à l’amélioration du pronostic des patients. Tout d’abord, l’adaptation de l’intensité des protocoles de chimiothérapies, définie individuellement, sur des critères cliniques et biologiques présents au diagnostic et sur la réponse précoce au traitement. On note également l’optimisation des soins de support, les progrès de la greffe de cellules souches hématopoïétiques et le développement d’alternatives thérapeutiques à la chimiothérapie, ciblant plus précisément les cellules tumorales (anticorps, cellules modifiées, petites molécules). Enfin, la mise en place d’essais cliniques et d’études collaboratives internationales a permis d’harmoniser les définitions, de standardiser les modalités d’évaluation et de comparer les schémas thérapeutiques utilisés par les différents groupes coopérateurs, permettant aussi d’améliorer la prise en charge des patients.
Les enjeux des prochaines années seront doubles : identifier précocement les patients à haut risque de rechute, éligibles aux nouvelles thérapies ciblées et sélectionner de façon toujours plus précise les patients au pronostic particulièrement favorable qui pourraient bénéficier d’un allègement thérapeutique afin d’éviter la survenue des effets secondaires inhérents à toute chimiothérapie.
Épidémiologie et mécanismes de développement des LAL
L’incidence annuelle des LAL aux États-Unis et dans les pays européens est relativement stable sur les dernières décennies avec environ 40-50 cas par million d’enfants et par an [1]. Ainsi, on dénombre chaque année environ 400 nouveaux cas de LAL chez les moins de 15 ans en France se répartissant pour environ 85 % d’entre elles en LAL de la lignée B (LAL-B) et 15 % en LAL de la lignée T (LAL-T). Le pic d’incidence se situe entre l’âge de deux et cinq ans [2].
De nombreuses études, menées à grande échelle, ont démontré l’impact de différents facteurs génétiques, environnementaux, infectieux ou immunologiques, à différentes périodes de la vie anté- et post-natale sur le développement des LAL [3].
En dehors de certaines altérations génétiques constitutionnelles rares, mais fortement associées à la survenue de LAL (ETV6, PAX5, IKZF1) ou de cancers incluant les LAL (TP53, gènes associés au syndrome CMMRD, ATM, trisomie 21, etc.), des études récentes utilisant des techniques de séquençage complet du génome (Genome Wide Association Studies – GWAS) ont mis en évidence des associations statistiques entre certains variants nucléotidiques impliqués dans la régulation transcriptionnelle ou la différenciation lymphoïde B et la survenue de LAL [4]. Ces polymorphismes sont retrouvés à une fréquence relativement importante dans la population générale mais ont une pénétrance faible et nécessitent donc probablement la coopération d’autres types de facteurs environnementaux ou l’association à d’autres facteurs de prédispositions génétiques.
Les liens de causalité entre les expositions environnementales et le risque de LAL ont été explorés par de nombreuses études. Cependant, peu d’entre elles ont pu être reproduites ou avoir la puissance statistique nécessaire pour démontrer avec certitude leurs hypothèses. À l’exception des radiations ionisantes, pour lesquelles ce sur-risque est prouvé, on retient comme facteurs de risque potentiels l’usage des pesticides au domicile, le tabagisme familial, la pollution de l’air et l’exposition professionnelle des parents aux solvants ou aux hydrocarbures [5].
Le système immunitaire semble en revanche avoir un rôle déterminant dans la survenue des LAL. La composition du microbiome des nouveau-nés et des nourrissons aurait un impact prolongé sur le fonctionnement lymphocytaire. Le défaut d’exposition aux agents microbiens dans la période néonatale et la petite enfance induit par l’hygiénisme des sociétés modernes, entraîne en effet chez les nourrissons et les enfants des perturbations immunitaires qui pourraient promouvoir le développement des LAL-B. Cette hypothèse est aujourd’hui soutenue par des études biologiques utilisant des modèles murins transgéniques pour des altérations génétiques déterminantes dans l’initiation des LAL (ETV6-RUNX1, PAX5, E2A-PBX1…). Le développement de LAL chez ces souris est directement influencé par la mise en contact avec les pathogènes de l’environnement. De plus, plusieurs variables épidémiologiques ou sociologiques liées indirectement à l’environnement microbien corroborent cette hypothèse. On retrouve ainsi une association entre LAL et la naissance par césarienne, l’absence de collectivité, l’allaitement artificiel, ou le rang de naissance bas, facteurs associés à une exposition microbienne moindre [6].
On sait depuis plusieurs années maintenant que les LAL suivent un processus de transformation multi-étapes pouvant prendre son origine in utero. Les preuves de cette initiation anténatale ont été notamment apportées par l’étude rétrospective des tests de Guthrie provenant d’enfants atteints de LAL avec translocation ETV6-RUNX1 [7] et MLL-AF4[8]. Les séquences génomiques de ces réarrangements étaient retrouvées chez la grande majorité des patients sur le sang néonatal et ce même chez des patients dont la LAL s’était développée longtemps après la naissance (jusqu’à l’âge de neuf ans). À l’inverse, la recherche systématique du transcrit ETV6-RUNX1 dans le sang de cordon placentaire d’une population de nouveau-nés non sélectionnés a permis de retrouver la présence de ce clone pré-tumoral chez 1 % des bébés soit plus de 100 fois la fréquence de ce sous-type leucémique chez l’enfant [9, 10]. Cela suggère donc que, si ces événements oncogéniques considérés comme initiateurs sont fréquents in utero, la majorité des clones pré-leucémiques ne progresseront pas vers une LAL, probablement du fait de l’absence d’altération génétique secondaire post-natale, qui constitue la seconde étape indispensable pour la transformation tumorale. L’étude des cas de LAL observées chez les jumeaux monozygotes a été un autre moyen de démontrer l’initiation fœtale des LAL pédiatriques. Il a en effet été possible, en suivant l’anomalie génétique initiatrice ou les séquences de recombinaison des gènes des immunoglobulines spécifiques de chaque LAL, de démontrer que des clones caractérisés par une même altération fondatrice pouvaient être retrouvés chez deux jumeaux mono-choriaux. À la faveur d’anastomoses placentaires, la cellule pré-leucémique apparue chez le premier jumeau est transfusée chez le second. Dans 10 à 15 % des cas environ, elle acquiert elle aussi des altérations génétiques additionnelles, la faisant progresser en cellule leucémique. Contrairement à l’anomalie initiatrice qui est commune, les événements secondaires apparaissent de façon indépendante dans la vie post-natale et sont donc différents entre les deux jumeaux. Dans le cas des LAL avec réarrangement du gène MLL, l’impact oncogénique du transcrit est suffisamment fort pour ne pas nécessiter d’événement coopérateur. Le risque de récurrence est alors de 100 % pour le second jumeau monozygote.
L’ensemble de ces études ont donc permis de mettre en évidence que le processus de leucémogenèse n’était pas dépendant d’un seul évènement causal mais était le résultat de l’interaction entre un terrain génétique constitutionnel particulier et divers événements environnementaux et génétiques secondaires pouvant survenir au cours du développement de l’hématopoïèse ante et post-natale.
Diagnostic clinique et biologique des LAL
Présentations cliniques
Les LAL sont des hémopathies malignes caractérisées par la prolifération de progéniteurs lymphoïdes bloqués à un stade précoce de leur différenciation. L’accumulation et l’expansion de ces cellules blastiques sont responsables de l’impossibilité pour les autres lignées hématopoïétiques de maturer de façon physiologique générant alors anémie, thrombopénie et neutropénie. Les signes cliniques relatifs à l’insuffisance médullaire constituent un mode de découverte fréquent de la pathologie (syndrome anémique, syndrome hémorragique et infection).
Associé à ce tableau d’insuffisance médullaire, les LAL se caractérisent parfois par l’existence d’un syndrome tumoral correspondant à une infiltration blastique hématopoïétique (adénopathies, hépato-splénomégalie, infiltrat thymique, douleurs osseuses) ou extra-hématopoïétique (rénale, testiculaire, cérébrale…). Ces atteintes sont parfois révélatrices de la maladie, et peuvent dans certains cas menacer le pronostic vital (compression médiastinale secondaire à l’infiltration thymique dans le cas des LAL-T par exemple). L’atteinte du système nerveux central (SNC) sera suspectée devant la présence d’un syndrome méningé, ou dans la majorité des cas au moment de la réalisation de la première ponction lombaire et/ou à la réalisation d’explorations complémentaires spécifiques (fond d’œil, imagerie cérébrale). À l’inverse, les atteintes testiculaires sont souvent diagnostiquées cliniquement (hypertrophie testiculaire pouvant être accompagnée de douleurs). Parmi l’ensemble de ces symptômes et localisations, seules les atteintes cérébrales et testiculaires [11] (environ 2 % et 5 % des cas respectivement) [12], plus fréquemment rencontrées dans les formes T, sont associées à un risque accru de rechute et imposent donc une intensification thérapeutique.
Enfin, l’hyperleucocytose en lien avec une expansion blastique périphérique parfois massive (8 % des patients atteints de LAL-B se présentent avec une leucocytose supérieure à 100 G/L mais pouvant atteindre jusqu’à 1 000 G/L ou plus) peut menacer le pronostic vital à court terme en raison des complications rhéologiques qu’elle génère (leucostase pulmonaire et cérébrale).
Il n’existe pas de signe clinique spécifique permettant de différencier les proliférations lymphoïdes B ou T. Cependant, on note au sein des LAL-T, un sex-ratio nettement en faveur des garçons (72 % de garçons dans les formes T vs. 54 % dans les formes B), un âge au diagnostic médian de neuf ans (vs. cinq ans pour les formes B), une leucocytose initiale > 100 G/L dans 40 % des cas (vs. 8 % pour les formes B) et dans plus de 30 % des cas une atteinte médiastinale pouvant menacer le pronostic vital [12, 13].
Hémogramme et biologie standard
Dans la quasi-totalité des cas, le diagnostic de LAL s’associe à des anomalies de l’hémogramme. Si environ la moitié des enfants se présentent avec une hyperleucocytose initiale (> 10 G/L), dans une minorité de cas aucun blaste n’est visible à la NFS et au frottis. L’anémie (Hb < 10 g/dL), normochrome, normocytaire, arégénérative est présente dans approximativement 80 % des cas au diagnostic. Sa profondeur est inversement corrélée à la rapidité d’installation de la prolifération tumorale. Dans les mêmes proportions, une thrombopénie (< 100 G/L) est également retrouvée. Enfin, on note parfois d’autres anomalies de la formule leucocytaire pouvant orienter vers certains sous types leucémiques particuliers comme l’hyperéosinophilie dans les LAL avec translocation t(5;14) (q31;q32),qui entraîne une élévation de la production d’interleukine 3, ou la myélémie dans les LAL acutisées de leucémies myéloïdes chroniques.
En dehors des perturbations de l’hémogramme, d’autres paramètres biologiques peuvent être perturbés au moment du diagnostic. Les LDH (lactates déshydrogénases) témoins de la « masse » tumorale, sont souvent augmentés sans qu’il s’agisse d’un signe spécifique de LAL. Le syndrome de lyse tumorale correspond au relargage de certains composés intracellulaires en conséquence de la destruction de cellules tumorales spontanée ou induite par le traitement. On observe alors une hyperphosphatémie associée à une hypocalcémie secondaire, une hyperkaliémie et une hyperuricémie. L’altération de la fonction rénale y est fréquemment associée du fait d’une précipitation de cristaux d’acide urique ou phospho-calciques. Elle peut être majorée par une infiltration blastique du parenchyme rénal. La présence d’une insuffisance rénale aiguë motivera une prise en charge rapide en milieu spécialisé et fera discuter rapidement le recours à des éventuelles méthodes de filtration extrarénale afin de prévenir les complications en lien avec le syndrome de lyse [14].
Diagnostic cytologique et immunophénotypique
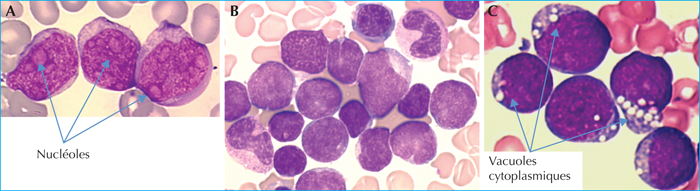
L’examen morphologique de la moelle osseuse réalisé sur des étalements médullaires colorés au May-Grünwald Giemsa (MGG) est un examen indispensable pour poser le diagnostic des LAL. Il permet de déterminer le pourcentage d’envahissement médullaire (par définition, une leucémie aiguë présente une infiltration médullaire blastique minimale de 20 %) [15](figure 1), d’analyser la morphologie des cellules tumorales (distinction lymphoïde/myéloïde, caractéristiques particulières de certains sous-types leucémiques) et de mettre en évidence des anomalies qualitatives d’autres lignées cellulaires pouvant orienter vers une pathologie sous-jacente (syndrome myélodysplasique, par exemple). Il permettait également auparavant de classer les LAL selon trois groupes (L1, L2 et L3 ; classification du groupe franco-américano-britannique (FAB)). Cette classification a été progressivement abandonnée au profit de classifications basées sur les caractéristiques immunophénotypiques, cytogénétiques et moléculaires.
La classification immunophénotypique des LAL (EGIL) [16] repose sur l’expression de marqueurs membranaires spécifiques de la maturation des cellules lymphoïdes dans la moelle ou le thymus tels que le CD10, CD19, le CD22 et le CD79a pour la lignée B ou le CD7, le CD3 intra-cytoplasmique et le CD5 pour la lignée T. Au sein des LAL-B, on distingue quatre formes principales :
- –Les formes immatures (ou pro-B) qui sont CD10 négatives et représentent moins de 5 % des formes de LAL-B des enfants de plus d’un an.
- –Les formes communes dites pré-pré B qui se caractérisent par la présence du marqueur membranaire CD10 et l’absence d’immunoglobuline de surface ou cytoplasmique.
- –Les formes pré-B qui sont CD10 positives et expriment en plus la chaîne cytoplasmique de l’immunoglobuline.
- –Les LAL B matures présentent une immunoglobuline de surface. Elles sont dites leucémies de Burkitt et représentent 2 % des patients et sont traitées comme les lymphomes de Burkitt.
Les LAL-T se répartissent en quatre sous-groupes (pro-T, pré-T, cortical et mature) selon le stade de différenciation. Ce degré de maturation cellulaire peut s’apprécier par l’expression des différents marqueurs T (CD1a, CD2, CD3, CD4, CD5, CD7 et CD8). On notera au sein des LAL-T immatures l’existence d’une forme particulière car associée à la présence de marqueur myéloïde et/ou de cellules souches hématopoïétiques, qualifiée d’ETP pour « Early Thymic Precursor » etreprésentant un peu plus de 10 % des LAL-T de l’enfant.
Les nouvelles techniques de cytométrie en flux (CMF) permettent la détection simultanée d’un grand nombre d’anticorps spécifiques, permettant d’individualiser avec une grande sensibilité le lymphoblaste. Ce principe de détection à la fois sensible et spécifique constitue également un outil de suivi de la maladie résiduelle.
Pièges diagnostiques
Malgré les progrès considérables dans la connaissance des LAL, l’utilisation des techniques les plus sophistiquées pour les comprendre, les suivre et les traiter, il paraît surprenant que ces pathologies puissent encore poser des problèmes diagnostiques. Pourtant, il existe plusieurs situations rares mais pouvant mettre en défaut la clinique et les premiers examens biologiques d’orientation.
Certaines LAL se présentent avec des douleurs ostéo-articulaires, à prédominance nocturne, associées à une fièvre et à un syndrome inflammatoire biologique pouvant ainsi mimer une arthrite juvénile idiopathique (AJI) ou une maladie de Still [17]. Ces patients présentent souvent une pancytopénie, sans blaste au frottis sanguin. La présence de facteurs anti-nucléaires, d’éruption cutanée, de signes objectifs d’arthrite peuvent quant à eux se manifester dans les deux maladies. L’analyse répétée de la NFS, du frottis sanguin et le myélogramme aident au diagnostic. Il paraît donc prudent de réaliser de façon systématique un myélogramme avant l’initiation d’une corticothérapie chez les patients chez qui une AJI est suspectée.
Les LAL peuvent être confondues avec d’autres pathologies responsables d’une dysfonction au minimum d’une des trois lignées hématopoïétiques. L’aplasie médullaire idiopathique ou plus rarement constitutionnelle peut se confondre au diagnostic avec une leucémie aiguë à présentation pancytopénique. C’est le myélogramme et parfois la biopsie médullaire qui permettront d’en faire la distinction [18]. Certaines LAL de présentation aplasique peuvent même avoir des phases de rémission spontanée de plusieurs mois avant d’évoluer vers des LAL [19], classiquement B. Le purpura thrombopénique idiopathique (et a fortiori les syndromes d’Evans) par la thrombopénie profonde et les signes hémorragiques associées peut également se confondre au diagnostic avec une leucémie aiguë, mais l’absence d’atteinte des autres lignées permettra d’éliminer une hémopathie maligne dans cette situation.
De nombreuses infections peuvent se présenter avec une symptomatologie proche de la LAL : EBV, CMV, rubéole, parvovirus, VIH, dengue, toxoplasmose, leishmanioses. Elles sont responsables de symptômes associant à des degrés variables cytopénies, adénopathies, hépato-splénomégalie et parfois activation macrophagique. La coqueluche peut également se manifester par une hyperlymphocytose avec de nombreux lymphocytes activés. De plus, dans un certain nombre de cas, la régénération médullaire induite à la suite d’infections, comme dans d’autres situations (sortie d’aplasie post-chimiothérapie, post-greffe…), entraîne dans la moelle osseuse une augmentation du nombre de précurseurs lymphoïdes B normaux, les hématogones. Ces cellules, dont le pourcentage peut aller jusqu’à 20-30 % peuvent être morphologiquement confondues avec les lymphoblastes. La CMF peut être d’une grande aide pour les distinguer des blastes, les hématogones ne présentant jamais d’anomalie de maturation.
Enfin, d’autres types de cancers pédiatriques peuvent être responsable d’un tableau d’insuffisance médullaire en lien avec une infiltration métastatique comme les neuroblastomes [20] ou les rhabdomyosarcomes alvéolaires [21].
Génétique des leucémies aiguës lymphoblastiques
Les LAL sont des pathologies clonales hétérogènes dont l’initiation est secondaire à l’apparition d’une altération génétique somatique pouvant survenir à différents stades du développement lymphoïde [3]. L’identification de ces événements génétiques récurrents, mutuellement exclusifs, est d’un intérêt capital pour la prise en charge des patients puisqu’elle a un impact direct sur le pronostic et la stratification thérapeutique. Le développement de techniques de biologie moléculaire au cours des dernières années a permis d’enrichir le spectre des altérations et l’on est aujourd’hui capable de retrouver une anomalie génétique « classante » chez plus de 90 % des enfants [22]. Elles concernent des translocations chromosomiques, des variations du nombre de copies (gain ou perte) et des mutations ponctuelles. La plupart de ces différents sous-groupes font l’objet d’un consensus dans la classification OMS [15]. De nouvelles entités parmi les LAL B, classées sous le terme « B-others » [23], ont été décrites plus récemment et font l’objet d’études afin de déterminer leurs mécanismes oncogéniques et leur pronostic.
Les nouvelles techniques de séquençage à haut débit de l’ADN et de l’ARN ont également révélé des altérations génétiques secondaires, nécessaires à la progression tumorale du clone fondateur. Elles peuvent être sous clonales, c’est-à-dire ne pas être retrouvées chez l’ensemble des cellules portant l’anomalie initiatrice. Elles peuvent être perdues ou sélectionnées au fil du traitement, sous l’influence des différents traitements ou à l’occasion d’une rechute. Les gènes mutés ou dérégulés le plus fréquemment sont impliqués dans le développement lymphoïde (PAX5, IKZF1…), la signalisation (CRLF2, IL7R, FLT3, NRAS, KRAS…), la régulation de la transcription (ETV6, ERG, TBL1XR1…), la régulation du cycle cellulaire (CDKN2A/CDKN2B, PTEN…), et la régulation épigénétique (CREBBP, EP300, SETD2…)[24]. Leur nombre, le type d’altération et les voies cellulaires ciblées sont étroitement corrélés avec l’altération initiale. Malgré leur caractère additionnel, ces anomalies peuvent radicalement changer le pronostic d’un patient et sont donc, elles aussi, utilisées dans la stratification thérapeutique.
Principales anomalies génétiques classantes des LAL de la lignée B (tableau 1)
Anomalies de bon pronostic
Ces sous-groupes génétiques sont majoritaires en pédiatrie et ont bénéficié au fil des protocoles d’une stratégie d’allègement thérapeutique. La translocation t(12;21) (p13;q22),récurrentedans les LAL-B pédiatriques (20 % des patients), génère un transcrit de fusion entre deux facteurs de transcription hématopoïétiques ETV6 et RUNX1 (anciennement TEL et AML1). Elle est cryptique au caryotype et est mise en évidence en FISH ou par recherche du transcrit de fusion en biologie moléculaire. Si ces formes sont associées à un pronostic particulièrement favorable, elles ont néanmoins la particularité de présenter de façon non exceptionnelle des formes de rechutes tardives voire très tardives [25]. Les LALhyperdiploïdes, c’est-à-dire ayant un nombre de chromosomes compris entre 51 et 65 par cellule, représentent environ 25 % des LAL de l’enfant. Les gains chromosomiques ne sont pas aléatoires et touchent préférentiellement les chromosomes 4, 10, 17 et 21 [26]. On note dans ce sous-type un enrichissement en mutations additionnelles touchant la voie RAS.
Anomalies de pronostic intermédiaire
La translocation t(1;19)(q23;13) impliqueles gènes TCF3 (anciennement E2A) et PBX1. Elle survient dans 5 % des LAL B de l’enfant. Son mauvais pronostic a été amélioré par la chimiothérapie intensive mais elle reste associée à un risque de rechute méningée [27]. Deux entités, récemment découvertes, semblent être associées à un risque intermédiaire et doivent donc recevoir un traitement par chimiothérapie intensive : les réarrangements de DUX4 qui induisent parfois une surexpression de ERG et les transcrits de fusion impliquant le gène ZNF384 [23].
Anomalies de mauvais pronostic
Ces patients gardent, malgré l’utilisation des protocoles de chimiothérapie les plus intensifs, des pronostics sombres. Ils bénéficient pour un certain nombre d’entre eux d’allogreffes de cellules souches hématopoïétiques et auront dans les prochaines années un recours de plus en plus important aux nouvelles thérapies ciblées dès la première ligne du traitement. La LAL-B avec translocation t(9;22) (ou à chromosome Philadelphie, Ph1) a la particularité d’être rare chez l’enfant alors qu’il s’agit de la forme la plus fréquente des LAL de l’adulte (3 % des enfants, 20 % des adultes et plus de 50 % des patients de plus de 50 ans). Elles entraînent la production du transcrit de fusion BCR-ABL1 qui code pour une protéine à activité tyrosine kinase. L’introduction de l’imatinib, un inhibiteur de cette activité tyrosine kinase, a permis d’améliorer la survie des malades [28]. Un sous-type de LAL B ayant un profil d’expression génique similaire à celui des LAL Ph1 mais sans transcrit de fusion BCR-ABL1 a été décrit il y a dix ans : les LAL BCR-ABL1-like ou Ph1-like[29, 30]. Plusieurs sous-groupes génétiques ont été identifiés au sein de cette entité. Les altérations qu’ils présentent entraînent une dérégulation de la signalisation dépendant des récepteurs aux cytokines et des tyrosines kinases. Certains d’entre eux peuvent bénéficier de traitements ciblés spécifiques (ITK, inhibiteurs de JAK…) [31, 32].
L’hypodipoïdie est définie par un nombre de chromosomes inférieur à 44. Cette entité représente 2 % des LAL-B pédiatriques etse divise en trois sous-groupes : « high hypodiploidy » (43 à 40 chromosomes), « low hypodiploidy » (entre 39 et 32 chromosomes), et « near haploidy » (entre 31 et 24 chromosomes) [33]. Ces sous-groupes présentent des profils d’expression et des mutations coopératrices différentes, celles ciblant la signalisation dépendante de RAS ou des récepteurs à tyrosine kinase étant associées au « near haploidy », celles ciblant le gène TP53 étant présentes chez plus de 90 % des patients de profil « low hypodiploidy », dont certaines à l’état constitutionnel (syndrome de Li-Fraumeni). LesLAL-B avec réarrangement du gène KMT2A (anciennement MLL) situé en 11q23 représentent seulement 5 % des LAL-B pédiatriques mais près de 80 % des LAL du nourrisson [34]. KMT2A code pour une histone méthyltransférase et régule la transcription de nombreux gènes, dont les gènes HOX impliqués dans la régulation de fonctions spécifiques des cellules souches hématopoïétiques. L’âge de développement très précoce, le faible taux de mutations additionnelles (essentiellement activant RAS et les tyrosines kinases), leur caractère sous-clonal et souvent non conservé à la rechute suggèrent le haut potentiel oncogénique du réarrangement de KMT2A.
D’autres anomalies sont associées à un pronostic sombre : l’amplification du gène RUNX1 ou iAMP21 [35], les translocations t(17;19) (q22;p13) entraînant la fusion TCF3-HLF, et plus récemment les réarrangements du gène MEF2D.
Anomalies génétiques des LAL de la lignée T
Les LAL-T présentent une oncogenèse complexe, au moins aussi diverse que les formes B [36]. Cependant, contrairement aux LAL-B, il n’existe pas de consensus actuel autour d’une classification oncogénétique stratifiante dans les formes T pédiatriques. Les LAL-T se caractérisent par un phénotype d’arrêt de maturation à un stade précoce au cours de la différenciation lymphoïde T. Ce stade d’arrêt de différenciation est corrélé avec la dérégulation d’oncogènes fondateurs et mutuellement exclusifs comme TLX1, TLX3, CALM-AF10 ou SIL-TAL1 [37].
En complément de ces altérations spécifiques d’un stade de maturation, ces leucémies présentent également de nombreuses dérégulations partagées par les différents sous-types de LAL-T et affectant différentes voies de transduction de signal intracellulaire, comme les altérations de la voie PI3 kinase-Akt (via les anomalies de PTEN) ou l’activation constitutive de la voie JAK-STAT ou encore les perturbations des gardiens du cycle cellulaire comme la perte de la protéine p16 (délétions du gène CDKN2A). Parmi ces anomalies, la plus fréquente des altérations concerne les perturbations affectant le récepteur transmembranaire NOTCH1, un facteur de transcription majeur impliqué dans la lymphopoïèse T. Les mutations de NOTCH1 (et/ou de son ubiquitine ligase FBXW7) sont retrouvées dans près de 70 % des LAL-T et semblent associées à un pronostic favorable chez l’adulte [38] comme chez l’enfant [39] à condition de ne pas être associées à des anomalies de la voie RAS et/ou PTEN.
Concernant les formes immatures de LAL-T dites ETP, ces dernières présentent une surreprésentation de certaines anomalies affectant des voies de régulation majeures (notamment la voie IL7/JAK-STAT et la voie des RAS/MAPkinase). Ce sous-groupe de LAL-T semble associé à un pronostic défavorable, en réalité l’impact sur la survie globale est controversé [40, 41] car vraisemblablement corrélée à l’intensité des protocoles de chimiothérapies.
Facteurs pronostiques
Afin de proposer aux patients un traitement d’intensité adapté à la sévérité de la leucémie, il est fondamental de déterminer le groupe de risque de chaque patient. La distinction de ces groupes était auparavant basée sur des facteurs pronostiques cliniques et biologiques simples : âge au diagnostic, leucocytose initiale et envahissement du système nerveux central. Le perfectionnement des techniques de cytogénétique et plus récemment de biologie moléculaire a permis d’affiner cette stratification qui repose actuellement sur de nombreux critères (tableau 1).
Facteurs liés à la présentation clinique
La caractérisation très précise des sous-groupes génétiques n’a pas effacé l’impact pronostique des caractéristiques cliniques du diagnostic. Ainsi, dans les LAL-B, un âge supérieur à dix ans (ou inférieur à un an) et/ou un taux de globules blancs supérieur à 50 000/mm3 sont considérés comme des facteurs stratifiants au diagnostic. Historiquement, ces deux seuils ont été fixés pour distinguer les LAL-B de risque standard (NCI-SR) des autres (NCI-HR) en sachant qu’environ deux tiers des patients se situent dans la première catégorie [42]. Les nourrissons de moins d’un an sont d’emblée considérés comme à haut risque et sont traités dans des protocoles spécifiques adaptés à leur âge mais également à leur risque cytogénétique particulier.
L’âge et le taux de GB ont en revanche un impact pronostique plus limité dans les formes T et ne sont pas stratifiants à ce jour bien qu’un taux de globules blancs supérieur à 200 000/mm3 semble prédictif de rechute dans ce sous-type de LAL [40].
Les atteintes neurologique ou testiculaire constituent également des facteurs de risque au diagnostic dans les LAL B et T. D’autres facteurs cliniques autrefois associés à un mauvais pronostic ont vu leur effet défavorable « disparaître » avec le perfectionnement des protocoles de chimiothérapies, c’est notamment le cas des formes de LAL avec une présentation « lymphomateuse » au diagnostic.
Facteurs liés à la caractérisation biologique et génétique
Comme décrit plus haut, les altérations génétiques initiatrices ont un impact fort sur le pronostic des patients. Leur proportion varie avec l’âge expliquant en partie l’aggravation du pronostic chez les enfants plus âgés chez qui les sous-groupes de bon pronostic sont quantitativement moins fréquents. Ainsi, au sein des LAL-B, les anomalies génétiques associées à un bon pronostic (hyperdiploïdie et translocation ETV6-RUNX1) représentent chez l’enfant 50 % des NCI-SR, 25 % des NCI-HR et seulement 8 % des adolescents et jeunes adultes [24].
Certaines altérations génétiques secondaires ont également un rôle dans le pronostic des patients. Les délétions d’IKZF1 sont détectées chez environ 15 % des patients mais cette proportion peut monter jusqu’à 70 % dans certains sous-groupes, comme les LAL Ph1 et Ph1-like[30]. Il a été démontré que leur présence grevait fortement le pronostic des patients, même chez ceux appartenant à un groupe de bon pronostic, à l’exception notable du groupe DUX4/ERG. L’impact d’autres délétions (CDKN2A, CDKN2B, PAR1…) est à l’étude et pourrait permettre d’affiner encore le pronostic de sous-groupes génétiques.
Facteurs liés à la réponse au traitement
Deux paramètres sont utilisés en pratique courante pour évaluer la réponse au traitement. Leurs modalités d’évaluation sont bien définies.
Cortico-sensibilité
La première évaluation est réalisée après une exposition à sept jours de prednisone et une ponction lombaire de méthotrexate. La cortico-sensibilité ou -résistance est définie par la présence d’un nombre de blastes circulants respectivement inférieur ou supérieur à 1 000/mm3 au 8e jour du traitement. Les formes cortico-résistantes représentent environ 10 % des LAL B [43] et 40 % des LAL T [39] et sont associées à un pronostic nettement plus défavorable, ce qui justifie une intensification thérapeutique chez ces patients. L’impact de ce facteur tend néanmoins à s’effacer devant l’évaluation de la maladie résiduelle en biologie moléculaire. Il n’est aujourd’hui plus décisionnel dans certains protocoles européens et américains.
Mesure de la maladie résiduelle ou MRD
Le délai nécessaire pour obtenir un taux de cellules leucémiques résiduelles indétectables est un des facteurs pronostiques majeurs dans les LAL de l’enfant. Ce délai de réponse est en réalité la conjugaison de nombreux facteurs indépendants mais potentiellement additifs tels que les caractéristiques cytogénétiques de la leucémie, le type et l’intensité du traitement reçu, ainsi que la pharmacocinétique et la pharmacodynamique propre à chaque patient (fonction de son âge et de ses polymorphismes génétiques).
Les techniques de biologie moléculaire actuelles permettent de détecter la présence d’une cellule leucémique au sein de 105 cellules hématopoïétiques normales. Ces techniques de mesure de maladie résiduelle (ou MRD pour Minimal Residual Disease) reposent sur la détection par PCR des réarrangements spécifiques du clone tumoral pour les gènes des immunoglobulines IGH ou TCR. La mesure de la MRD peut également se réaliser par CMF via la recherche de l’expression aberrante d’une combinaison de marqueurs au diagnostic et au cours du traitement.
Les études récentes retrouvent un risque de décès trois à cinq fois supérieur parmi les enfants avec une mesure de la MRD positive par rapport aux enfants avec une MRD négative [44]. Ainsi, une MRD dont l’évolution ne serait pas satisfaisante peut justifier à elle seule une indication d’allogreffe chez des patients en première rémission. Si l’impact pronostique que représente ce marqueur est aujourd’hui admis internationalement, il persiste en revanche des interrogations sur le meilleur moment pour interpréter la qualité de cette réponse post-traitement. Ce constat est particulièrement vrai pour les formes T où certaines études mettent en évidence une plus lente décroissance de la MRD au cours du traitement, sans impact pronostique péjoratif. Pour ces auteurs, c’est la mesure de la MRD « tardive » (post-consolidation au lieu de post-induction) dans les formes T qui aurait un réel impact pronostique [45]. Répondre à cette question centrale puisque source de modification thérapeutique majeure, est notamment un des enjeux des protocoles thérapeutiques actuels.
Les grands principes du traitement des LAL
Les premiers succès thérapeutiques
Depuis la première description en 1948 par Sidney Farber [46] des premières rémissions, transitoires, de LAL grâce à un traitement reposant sur un anti-folates ; l’aminoptérine, des progrès thérapeutiques exceptionnels ont été réalisés depuis, notamment via l’émergence des schémas de polychimiothérapies au cours des années 60. Ces premières combinaisons ont permis d’obtenir des rémissions complètes dans près de 80 à 90 % des cas [47]. Cependant, ces enfants présentaient une rechute dans près de 75 % des cas, à point de départ neuroméningé, puis secondairement médullaire. L’addition de ponctions lombaires avec injections intrathécales de chimiothérapie et l’ajout d’une irradiation cranio-spinale dans le schéma protocolaire de traitement des LAL ont permis de faire chuter drastiquement le nombre de rechute neuro-méningée et d’augmenter les taux de survie des LAL (figure 2) [48].
Un autre tournant majeur dans le traitement des LAL a été l’introduction par le groupe Berlin-Frankfurt-Münster (BFM) d’un schéma toujours utilisé aujourd’hui reposant sur l’utilisation de huit médicaments pendant huit semaines (induction puis consolidation) [49]. Ce protocole constitue encore aujourd’hui la colonne vertébrale de la quasi-totalité des protocoles internationaux de traitement des LAL (adultes et pédiatriques).
Principes des protocoles actuels de chimiothérapie
La trame actuelle de traitement des leucémies aiguës lymphoblastiques repose sur la succession de plusieurs phases de chimiothérapies :
- –l’induction (trois à cinq molécules) dont l’objectif est l’obtention de la rémission complète (définie par un examen clinique normal, la reprise d’une hématopoïèse normale attestée par une NFS normale et une blastose médullaire inférieure à 5 % à J35-J42) ;
- –la consolidation utilisant principalement des molécules non administrées en induction pendant environ huit semaines ;
- –l’intensification retardée pour laquelle est utilisée une polychimiothérapie reprenant notamment les molécules de l’induction. Le bénéfice d’une intensification tardive (ou de deux en cas de facteur pronostique péjoratif) a été démontré, y compris pour les LAL de risque standard. Dans les LAL de risque intermédiaire ou de haut risque, respectivement une ou deux phases comportant des injections de méthotrexate à haute dose y sont associées ;
- –le traitement d’entretien dont l’objectif est l’éradication d’éventuelles cellules blastiques résiduelles. Le traitement d’entretien est principalement ambulatoire. Il associe l’administration orale quotidienne de 6-mercaptopurine et hebdomadaire de méthotrexate pendant 18 à 24 mois. La première année s’ajoutent éventuellement des réinductions mensuelles intraveineuses de vincristine associées à une corticothérapie de courte durée. La prophylaxie méningée par injections intrathécales trimestrielles se poursuit pendant cette phase.
La chimiothérapie systémique ne diffusant pas toujours suffisamment à travers la barrière hémato-méningée afin d’assurer une prophylaxie neuroméningée efficace, des injections intrathécales de chimiothérapie sont réalisées tout au long du traitement pour prévenir les rechutes du système nerveux central. Son intensité sera fonction de l’atteinte initiale du SNC et/ou du risque de rechute neuroméningée estimée par différentes caractéristiques propres à la LAL. Pour les patients à très haut risque de rechute méningée, l’administration de perfusions de méthotrexate haute dose peut être préconisée.
La transplantation de cellules souches hématopoïétiques en première rémission complète est réservée aux LAL de haut risque soit moins de 10 % des enfants : ceux ayant des formes génétiques de très mauvais pronostic et surtout chez ceux ayant une décroissance non optimale de leur niveau de MRD [50].
Conclusion
L’optimisation des combinaisons de chimiothérapies, l’amélioration des soins de support, le développement de l’allogreffe de cellules souches hématopoïétiques et la performance du diagnostic couplés au suivi biologique et immunologique des patients, ont permis, au cours des dernières décennies, de faire décroître drastiquement la mortalité des patients traités pour une LAL. Il reste cependant de nombreux défis à relever. Les objectifs des années à venir seront à la fois de diminuer le recours aux chimiothérapies potentiellement sources de séquelles à long terme des patients présentant les leucémies dont le pronostic est le plus favorable mais également d’améliorer le pronostic des patients atteints des formes agressives de leucémies résistantes aux chimiothérapies classiques. Pour cela, les cliniciens devront prendre en compte d’une part les nouveaux outils biologiques de caractérisation des cellules cancéreuses, intégrer le métabolisme et le système immunitaire propre au patient et d’autre part s’appuyer sur les nouvelles alternatives thérapeutiques à la chimiothérapie actuellement en développement. Notamment via l’inhibition des mécanismes oncogéniques complexes en ciblant les molécules de surface ou encore via la modulation de la réponse immunitaire, qui seront autant de nouvelles armes thérapeutiques à la fois plus précises et plus efficaces dans le ciblage des cellules tumorales et l’épargne des cellules saines.Points à retenir
Liens d’intérêts
les auteurs déclarent n’avoir aucun lien d’intérêt en rapport avec l’article.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International