Epileptic Disorders
MENUTrisomy 20p/monosomy 18p associated with congenital bilateral perisylvian syndrome Volume 24, numéro 3, June 2022

Polymicrogyria (PMG) is a malformation of cortical development (MCD) characterized by excessive gyration and abnormal cortical layering [1]. Bilateral perisylvian polymicrogyria (BPP) is the most common subtype of polymicrogyria [1]. The association between BPP andoro-motordysfunction, epilepsyand cognitive impairment defines a distinct clinical condition named “congenital bilateral perysilvian syndrome” (CBPS) [2]. Genetic causes are collectively rare for BPP and typically occur in clinically recognizable syndromic forms, the most common of which are 1p36.3 and 22q11.2 deletion syndromes. Recently, Stutterd et al. reported a monogenic cause of PMG in up to 20% of patients [3].
Monosomy of the short arm of chromosome 18 (18p-) and trisomy of the short arm of chromosome 20 (20p+) are well-known chromosomal aberrations. 18p- is among the most frequent life-compatible monosomies in humans [4]; 20p+ is rarer but is also compatible with life in the majority of cases [5]. Most cases of 20p+ result from reciprocal translocations [5]. Despite the ever-growing numbers of affected subjects, the unique combination of these two chromosomal aberrations has been previously reported in only three subjects, featuring overlapping symptoms including severe neurodevelopmental delay, hypotonia and dysmorphic features [6-8]. To date, neither malformation of cortical development (MCD) nor epilepsy have been associated with trisomy 20/monosomy 18. Here, we report the first subject presenting with CBPS associated with unbalanced translocation resulting in partial monosomy 18p and partial trisomy 20q.
Case study
This 29-year-old Danish male was born at term with normal delivery, after a pregnancy complicated by pre-eclampsia. At three months of life, overt craniofacial dysmorphisms were noticed such as low-set ears, micrognathia, anteverted nostrils, short neck, broad hands with brachydactyly, proximal displacement of the thumbs, and flat feet with over-riding toes. Karyotyping showed paternally inherited 46,XY,der (18),t(18;20)(p10;q10)pat, as reported in a previous publication [9]. Segregation analysis revealed a balanced reciprocal translocation in nine healthy family members. The patient’s mother had several miscarriages for females all carrying the balanced translocation. No genetic testing was performed on the foetuses.
In the first year of life, the subject displayed neurodevelopmental delay, hypotonia, fatigue, and episodes of motor arrest without EEG correlates. A CT scan showed cerebellar atrophy and a widened left fissura Sylvii. Echocardiography revealed an atrial septal defect, which closed spontaneously at follow-up. At 18 months of age, he started to suffer from brief tonic asymmetric seizures during wakefulness and sleep that occasionally evolved to bilateral tonic seizures, refractory to antiseizure medication. In the following years, he developed chronic renal failure and hypertension due to congenital renal dysplasia. He underwent kidney transplantations at 16 and 18 years. Since the age of 24 years, he has been followed at the Danish Epilepsy Centre.
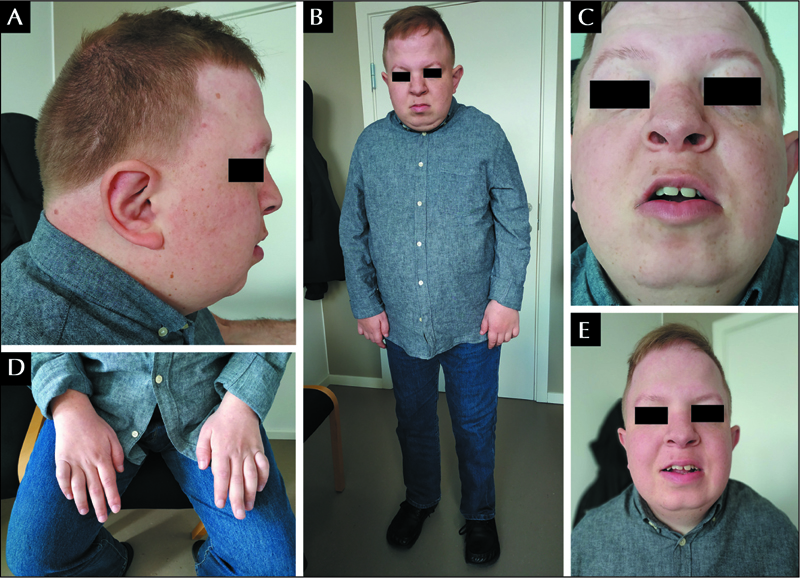
At last examination, at the age of 29 years, he displayed moderate-to-severe intellectual disability. He could understand several simple and a few complex messages. His vocabulary was poor and he primarily communicated using body and sign language. He presented with dysmorphic features, which are illustrated in figure 1A, C, E. He had short stature (160 cm; SD: -3.5), a broad chest with sloping shoulders and dorsal kyphosis, rhizomelic shortening of the extremities with broad and short hands, and brachydactyly. He showed diffuse hyperpigmented spots on the skin. His sight was severely impaired due to optic nerve hypoplasia and divergent strabismus. The protrusion as well as the lateral movements of the tongue were extremely limited, consistent with oro-motor impairment. Simple motor tasks on request were performed slowly and with a delay. Impairment of both gross and fine motor functions were evident; gait was unstable and bradykinetic, but possible without support. He showed a severe limitation in flexion, extension and rotation of the neck and his body posture was characterized by standing with a mildly wide base and leaning slightly forward (figure 1B). Proximal strength was conserved and distal strength was reduced. Deep tendon reflexes were hyperexcitable in lower limbs and normal in the upper limbs. The forearms were pronated and flexed, and proximal interphalangeal joints of bilateral fourth and fifth fingers were contracted (figure 1D). He suffered from chronic constipation.
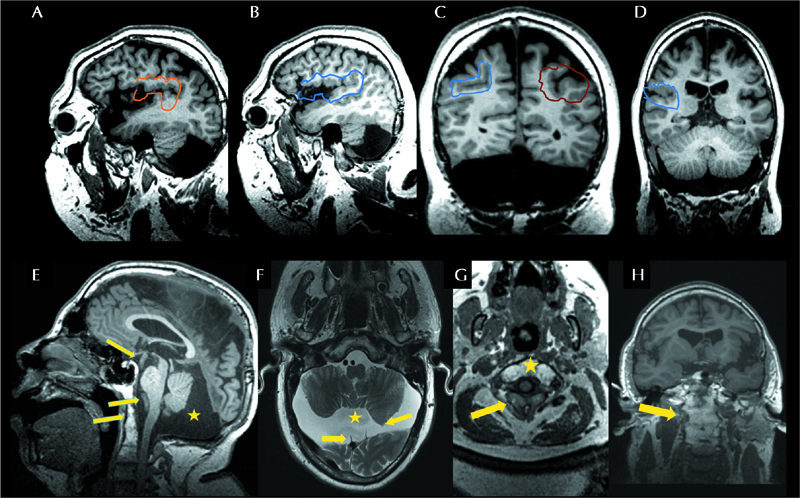
3-Tesla brain MRI, performed at the age of 28 years, showed a complex brain malformation that included microcephaly with sloping of the forehead, enlarged lateral ventricles and wide sulci and PMG on both temporal lobes, predominantly on the left side and on the right fronto-parietal operculum, consistent with BPP (figure 2A-D). A hypoplastic corpus callosum, hypoplasia of the cerebellum, a small neuroglial cyst in the right parietal lobe, a large mega cisterna, scalloping of the occipital bone, and tortuosity of brain arteries with dolicoectasia of the larger arteries were also detected. In addition, a vertebral malformation, characterized by non-segmentation of anterior C1-C2 and fusion of posterior elements with a bifid spinous process, a narrow spinal canal and probable spinal stenosis of C2-C3, were observed (figure 2E-H). He suffered from weekly epileptic tonic seizures despite polytherapy with lacosamide, lamotrigine and rufinamide. During long-term EEG recording, interictal EEG showed left-sided spike-waves/sharp waves with posterior predominance (supplementary figure 1A). Ictal EEG during asymmetric tonic seizures with onset on the left arm showed rhythmic 5-6-Hz activity arising from the right fronto-central region and spreading to the right posterior region, to the midline and to the left centro-fronto-temporal regions (supplementary figure 1B). Electrical source imaging (BESA Research software 6.1) of the ictal EEG activity showed a perisylvian source of the epileptic activity in the posterior part of the right temporal operculum, close to the posterior part of the insula (supplementary figure 1C-E).
The array-CGH confirmed an unbalanced translocation with a deletion of the whole short arm of chromosome 18 and a duplication of the whole short arm of chromosome 20 (arr(18p)x1, (20p)x3; 400K oligoarray; minimal resolution 200 kb).
Discussion
Our report documents the association between CBPS and unbalanced translocation trisomy 20p/monosomy 18p, which has not previously been reported. Indeed, clinical signs, such as intellectual disability, drugresistant epilepsy with tonic seizures, oro-motor dysfunction and craniofacial dysmorphisms, together with the MRI evidence of BPP, are distinctive features of CBPS [1, 2]. Additional non-cortical malformations observed in our patient, such as hypoplasia of the corpus callosum and cerebellum, have been previously reported in patients featuring PMG [1].
In the literature, cerebral malformations associated with 20p+ are rare. One case of anencephaly has been described [10] but no subjects with 20p+ have been reported to have MCDs. Anecdotal associations between 18p- and holoprosencephaly and/or defects of the midline, such as an absent olfactory tract and bulbs, corpus callosum agenesis and hypopituitarism, as well as Dandy Walker malformation with hydrocephalus, perisylvian PMG and a mild cerebellar vermis hypoplasia, have been reported [4, 11]. These findings have suggested a possible locus associated with these malformations, including PMG, on the short arm of chromosome 18 [11], however, known MCD-associated genes have not been located to chromosome 18 [12]. A paper from Alkan and colleagues [13] reported a patient affected by partial monosomy of both 21q and 18p with a neuronal migration disorder, however, no other similar cases have been reported in the literature. More often, trisomies and tetrasomies of 18p [14] have been described in association with MCD, but still, the genes associated with these malformations are unknown.
These data suggest that patients with PMG should be screened using array-CGH and karyotyping, even with a unilateral or focal form of PMG. Array-CGH should also be used to identify breakpoints and regions containing putative genes involved in brain development, the pathogenic variants of which might be associated with MCD. BPP has never been reported previously in patents with 18pand 20p+. This might depend on several factors including reduced penetrance, difficulties in performing MRI investigations without sedation, or insufficient quality of MRI scanning in the earliest reports. Lastly, reported cases often feature partial and incomplete deletion/duplications of the chromosome. This may suggest that aberrations do not affect loci that are critical for the development of the malformation and could be proximal to the point of rupture.
Epilepsy is rarely associated with 18p- or 20p+ [15], but has been reported in up to 87% of individuals with CBPS [2]. Seizure semiology is stereotyped over the years, and is characterized by tonic motor manifestations with EEG findings indicating involvement of perisylvian polymicrogyric areas, as previously reported in CBPS [2].
A peculiar disturbance in our subject was the severe impairment of head and neck motility leading to an abnormal posture. A posture with sloping shoulders and the trunk leaning forward has been suggested as typical of 18-[4], however, in our case, the role played by the fusion of cervical vertebrae should be considered. Other features present in our subject, such as the cardiac defects and renal abnormalities, are seen in 20p+ [5], whereas the 18p- may account for the short stature, abnormal hands and feet, and bradykinesia. Facial gestalt includes dysmorphic features associated with both chromosomes leading to a typical 18p-/20p+ facial gestalt, as previously proposed [8].
In conclusion, our report describes, for the first time, the association between 18p-/20p+ and CBPS, also illustrating the course of this condition into adulthood, thus providing useful information to better counsel families about the developmental prognosis of this rare unbalanced chromosomal rearrangement.
Supplementary material.
Supplementary figure accompanying the manuscript is available at www.epilepticdisorders.com.
- (1)Which is the definition of polymicrogyria?
- Polymicrogyria is a malformation of cortical development characterized by excessive gyration and abnormal cortical layering.
- Polymicrogyria is a malformation of cortical development characterized by abnormal transmantle neuronal migration.
- Polymicrogyria is a malformation of cortical development characterized by a lack or reduction of gyration.
- (2)Which features characterize the “congenital bilateral perisylvian syndrome” (CBPS)?
- Bilateral perisylvian polymicrogyria, pyramidal signs, epilepsy and cognitive impairment.
- Bilateral perisylvian polymicrogyria, oro-motor dysfunction, epilepsy and cognitive impairment.
- Frontal polymicrogyria, movement disorder, epilepsy and behavioural issues.
- (3)Which chromosomes are most frequently affected in patients with bilateral perisylvian polymicrogyria?
- Chromosomes 18 and 20.
- Chromosomes 1 and 22.
- Chromosomes 21 and 1.
Acknowledgements and disclosures.
We thank the patient and his parents for their kind cooperation. None of the authors have any conflicts of interest to disclose.