Epileptic Disorders
MENUBenign familial infantile epilepsy associated with KCNQ3 mutation: a rare occurrence or an underestimated event? Volume 22, numéro 6, December 2020

Illustrations
- Mots-clés : benign familial infantile epilepsy, BFIE, KCNQ3, mutation, incidence
- DOI : 10.1684/epd.2020.1221
- Page(s) : 807-10
- Année de parution : 2020
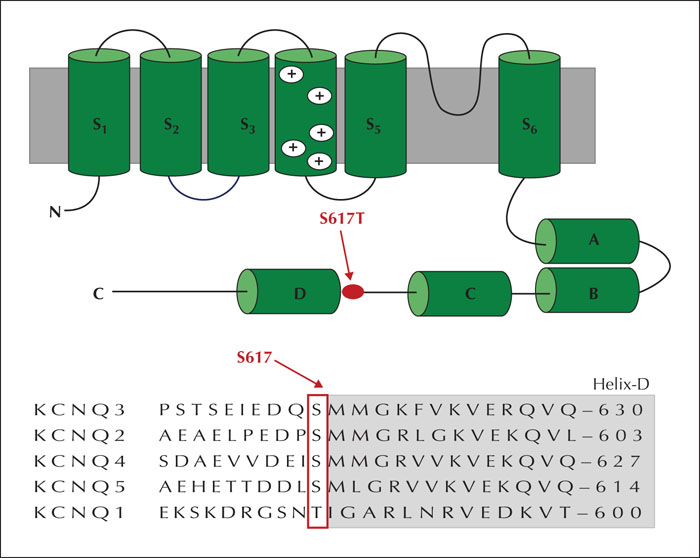
Benign familial infantile epilepsy (BFIE) is the most genetically heterogeneous phenotype among early-onset familial infantile epilepsies. It has an autosomal dominant inheritance pattern with incomplete penetrance. Although PRRT2 is the most mutated gene detected in families with BFIE, other mutations in KCNQ2, SCN2A, and GABRA6 genes have also been described. To date, KCNQ3 mutations have been detected in only four patients with BFIE. Here, we describe the clinical pattern and course of an additional individual with BFIE associated with a novel missense heterozygous KCNQ3 c.1850G>C variant inherited by his unaffected father. The incidence of KCNQ3 mutations among BFIE patients is reported to be low in the literature, however, whether this is underestimated is unclear as not all current epilepsy gene panels include KCNQ3.