Epileptic Disorders
MENUAntiepileptic drugs: evolution of our knowledge and changes in drug trials Volume 21, numéro 4, August 2019

“On the 20th of May 1747, I took twelve patients in the scurvy, on board the Salisbury at sea. Their cases were as similar as I could have them… They lay together in one place… and had one diet common to all. Two of these were ordered each a quart of cyder a day. Two others took twenty-five drops of elixir vitriol three times a day… Two others took two spoonfuls of vinegar three times a day… Two… were put under a course of sea water… Two others had each two oranges and one lemon given to them every day… The two remaining patients took …an electary recommended by an hospital-surgeon, made of garlic, mustard feed, rad. raphan. balsam of Peru and gum myrrh… The consequence was, that the most sudden and visible effects were perceived from the use of the oranges and lemons; one of those who had taken them, being at the end of the six days fit for duty… The other was the best recovered of any for this condition; and being now deemed pretty well was appointed nurse to the rest of the sick.”
James Lind MD. A treatise on the scurvy. London : Printed by A. Millar in the strand, 1757 : 140-50.
Although the first implementation of the randomized controlled trial dates back to the 18th century, its use to evaluate the efficacy and safety of medicines became established only 200 years later, with the publication of the iconic MRC trial of streptomycin for the treatment of tuberculosis (Medical Research Council, 1948). In fact, the medications used in the treatment of epilepsy in the first half of the 20th century were introduced in the market without any kind of controlled assessment. Admittedly, during that period, effective treatments were introduced that remain part of the modern armamentarium (bromides, phenobarbital, phenytoin, and the ketogenic diet), but lack of rigorous scientific assessment also led to utilization of many agents of questionable value. A standard British textbook published in 1940 listed borax, belladonna, and nitroglycerine among drugs of definite benefit for the treatment of epilepsy (Kinnear-Wilson, 1940), while an article by Lennox (1940) listed not only phenobarbitone and phenytoin, but also brilliant vital red, borotartrate, and antirabies vaccine among epilepsy treatments used in America in the mid-30s.
The evolution of the methodology for the clinical assessment of potential antiepileptic drugs (AEDs) is a fascinating history, which also bears witness to advances in our understanding of these drugs and therapeutic achievements (Arzimanoglou et al., 2010). This article provides an overview of clinical trials and advances in knowledge since 1938 – the year of introduction of phenytoin and also the year in which evidence of safety was made a requirement for the marketing of medicines in the US (table 1). Interestingly, demonstration of efficacy was made a requirement only in 1962 in the US, and several years later in Europe.
The age of the pioneers (1938-1969)
The discovery of phenytoin and its legacy
The introduction of phenytoin by Merritt and Putnam can be regarded as the beginning of modern AED development. In fact, Merritt and Putnam's studies have been described as ‘monumental landmarks in the history of epilepsy, pharmacology, and neurology’ (Rowland, 1982). This is not only because the clinical value of phenytoin at that time was truly ‘revolutionary’ (Lennox, 1940), but also because the methodology applied to the development of phenytoin (preclinical screening in a seizure model, e.g. electrically induced convulsions, followed by clinical testing) applied a paradigm that continues to be used to date (Friedlander, 1986). Yet, in terms of clinical trial methodology, the development of phenytoin can hardly be described as exemplary. Shortly after completing their laboratory experiments towards the end of 1936, Merritt and Putnam started trying the compound in 200 patients with uncontrolled seizures and by June 1938 they were ready to present their findings at the section on Nervous and Mental Diseases of the American Medical Association (AMA) (Merritt and Putnam, 1938). Based on their observations, they concluded that phenytoin ‘was effective in controlling convulsive seizures in a great majority of a selected group of patients who were not helped by other methods of therapy’ even though, perhaps surprisingly, the drug was considered ‘more toxic than bromides and the barbituric compounds’. In the same month, phenytoin (Dilantin) was added to the catalogue price list of Parke-Davis, as there was no requirement for a license by a regulatory authority at that time (table 1). So, it took only two years from the first experiments in animal models to the marketing of the drug, and the clinical development program lasted about one year, without any controlled study. Although it is unclear how many patients had been exposed at the time phenytoin was marketed, a review of the evidence made in late 1939 by the AMA Council on Pharmacy and Chemistry listed 13 clinical studies with a total of 595 patients, including the original series reported by Merritt and Putnam (Council on Pharmacy and Chemistry, 1939). Based on these data, phenytoin was included in the Council's New and Nonofficial Remedies, with the indication for use in ‘epileptic patients who are not benefited by phenobarbital or bromides and in those in whom these drugs induced disagreeable side reactions’. This restrictive indication, perhaps surprising based on the value of the drug, is reminiscent of the cautious labeling of some AEDs introduced many decades later under totally different regulatory scenarios.
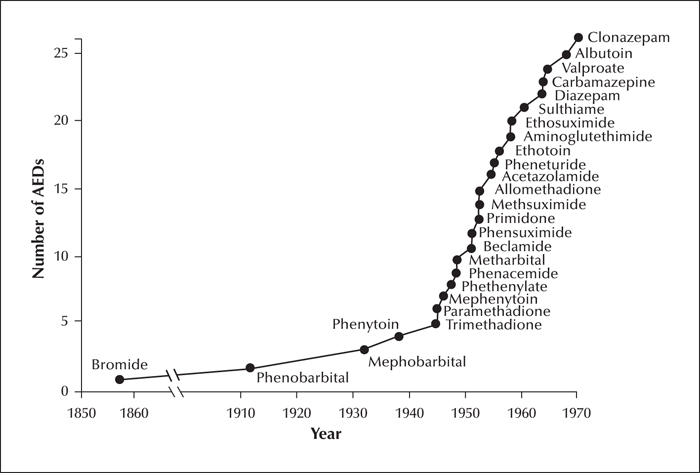
The discovery of phenytoin left an important legacy for AED development. First, it demonstrated that, contrary to beliefs widely held at that time, antiseizure activity can be obtained in the absence of sedative effects, thereby stimulating the pharmaceutical industry to search for similarly innovative compounds. Second, the electrical stimulation test used by Merritt and Putnam provided a screening tool for AED discovery, and an incentive to develop improved models such as seizures induced by maximal electroshock and chemoconvulsants. Third, the hydantoin molecule offered the opportunity to explore the activity of various structurally related compounds, which went on to be developed as AEDs. The number of AEDs that came to the market during the 30 years following the introduction of phenytoin (figure 1) is impressive and resembles the surge of activity that characterized the licensing of second-generation AEDs in the last three decades. Overall, the age of pioneers saw the development of many AEDs that remain widely used today, including carbamazepine, phenytoin, valproate, ethosuximide, and benzodiazepines. Admittedly, many AEDs marketed in that period did not withstand the test of time and have been withdrawn from the market or are rarely used today. This is likely to reflect to some extent the lack of rigorous requirements for the marketing of medicines in those years, which resulted in many compounds being introduced without adequate clinical trials. During the age of pioneers, application of the randomized controlled trial to epilepsy studies was just in its infancy, as discussed in the next section.
Clinical trials during the age of the pioneers
The state of the art of clinical trials during these years is best summarized by the report by Coatsworth (1971), who surveyed published studies on the efficacy of the AEDs available at that time. The report highlighted major methodological deficiencies. Of 110 clinical trials which were closely assessed, only three used an adequate control for bias, and only two had a double-blind design. Additionally, many trials were underpowered, had short evaluation periods, and lacked a precise description of the type of epilepsy studied. This was not helped by the fact that a widely accepted and well-structured classification of seizures and epilepsies did not become available until 1969 (Gastaut, 1969a, 1969b).
An interesting example of trial methodology in the 60s is the study of Gruber et al. (1962), who applied a double-blind crossover design to compare seven different AEDs, each at three different doses, with placebo in 44 patients with epilepsy, mostly associated with focal brain damage. Each testing period lasted five days (Tuesday through Saturday) and the patients’ usual medications were discontinued during these periods. The practice of discontinuing concomitant treatment when testing AEDs was not uncommon at that time, but by modern standards would be considered ethically problematic (particularly so in this study, in view of the low drug doses tested) and may have introduced a confounder by precipitating withdrawal seizures. By calculating seizure scores based on number of seizures during each observation period, the authors determined that the following daily doses of each drug could be considered equivalent in controlling motor seizures: phenobarbital 30 mg, phenytoin 93 mg, metharbital 110 mg, mephenytoin 112 mg, methylphenobarbital 128 mg, and primidone 200 mg. Ethotoin was poorly effective and 470 mg/day of this drug was considered to be equivalent to 70 mg/day phenytoin. One of the main limitations of the study was the short evaluation period for each treatment, which was inadequate to characterize accurately the seizure response and did not permit achievement of steady-state serum concentrations for most treatments. Carry-over effects from the test drugs and from the patients’ usual medications, which were reintroduced during each two-day interval between testing periods, could also have confounded results. Although the authors acknowledged some of these limitations, this trial illustrates how clinical assessment of AEDs in the early days was hampered by methodological constraints.
Another interesting randomized double-blind trial in the same period evaluated the comparative efficacy of primidone, phenobarbital, phenytoin, and their combinations in a total of 20 patients with focal seizures (White et al., 1966). By using a 10 × 10 Latin square design, 10 different treatment schedules, each given for two-week periods without wash-out, were compared: phenytoin 300 and 600 mg/day, phenobarbital 150 and 300 mg/day, primidone 750 and 1,500 mg/day, phenytoin 300 mg/day and phenobarbital 150 mg/day, phenytoin 300 mg/day and primidone 750 mg/day, phenobarbital 150 mg/day and primidone 750 mg/day, and placebo. During the study, rescue medication with 200 mg phenobarbital orally was given if a seizure occurred in any eight-hour interval, with another 200 mg phenobarbital dose added if a second seizure occurred in the same interval. If a third seizure occurred, an enema, followed by a suppository containing 100 mg amobarbital sodium, was given. Efficacy was assessed by calculating ‘demerit points’ according to number of seizures during each testing period, with additional demerit points given whenever a patient had to be withdrawn from a study period due to excessive seizures or toxicity. The trial showed a trend for phenytoin to have greater antiseizure activity than the other drugs, and for each drug, the higher (double) dose was more efficacious than the lower dose. The efficacity of combination therapies was roughly equal to twice the large dose of either agent included in the combination. In view of the doses used in the high-dose groups, it is remarkable that only five patients were withdrawn from a study period due to toxicity. Details of adverse events, however, were surprisingly not reported. Although conceptually stimulating, particularly regarding the comparison of single-drug therapies with drug combinations, this study also shows many of the limitations discussed for the previous example, including short evaluation periods and the potential influence of carryover effects and other confounders.
The hiatus (1970-1988)
‘The ‘hiatus’ refers to a period during which no major additional AEDs were introduced. This long interval justifies the commonly used differentiation between ‘old’ and ‘new’ generation drugs. The hiatus, however, was a fruitful period for epilepsy research, and led to important therapeutic advances.
Benefits of a better understanding of the properties of existing AEDs
The 70s and the 80s saw the flourishing of investigations on the clinical pharmacology of AEDs, including many well designed randomized controlled trials (discussed in the next section). These studies were crucial to:
- –characterize the pharmacokinetics and drug interactions of available AEDs, and their relationship with clinical response (Richens, 1980; Perucca, 1982; Perucca and Crema, 1983);
- –improve our understanding of the spectrum of activity of AEDs in different seizure types, including the possibility of AED-induced seizure aggravation (Shields and Saslow, 1983);
- –characterize the acute and chronic adverse effects of AEDs (Reynolds, 1975; Reynolds and Trimble, 1985), and provide comparative effectiveness data to guide drug selection and use.
Ultimately, these studies led to establish the basic principle of AED therapy, i.e. the tailoring of drug selection and dose to the characteristics of the individual. Admittedly, the knowledge acquired during that period had many important gaps, including very limited information on comparative risks of foetal AED exposure in the era preceding the establishment of AED and pregnancy registries. Nevertheless, strong signals for a higher teratogenic risk of valproate emerged as early as 1982 (Robert and Guibaud, 1982).
An important component of advances in quality of care during this period came from increased application of pharmacokinetic principles and therapeutic drug monitoring (TDM) to the individualization of dose. Interest in this area originated from early work of Fritz Buchtal in Copenhagen (Schiller and Buchtal, 1958; Schiller and Buchtal, 1967; Buchtal and Svensmark, 1971) and Lars Lund in Stockholm (Lund, 1974), and was advanced by the studies of Alan Richens in Europe (Richens and Dunlop, 1975) and Charles Pippenger in the US (Pippenger et al., 1976). A related landmark development in the late 70s originated from the work of Ted Reynolds and Simon Shorvon in London. Utilization of polypharmacy, largely prevalent at that time, was recognized as being often associated with undue toxicity without necessarily improving seizure control (Shorvon and Reynolds, 1977). By contrast, most patients could be optimally controlled with a single drug, as shown by two prospective studies in individuals newly diagnosed with focal and/or tonic-clonic seizures (Shorvon et al., 1978). One-drug therapy with either phenytoin or carbamazepine, assisted by serum drug level monitoring, resulted in seizure freedom rates of 76-88%. Although by modern standards these studies were methodologically weak (an uncontrolled design was used, and the number of patients included was only 31 in the phenytoin study and 25 in the carbamazepine study), their findings were highly influential. In fact, the message that most patients achieve seizure freedom on the initially prescribed AED has been repeatedly confirmed in subsequent studies, even though response rates may not be as high as originally reported (Kwan and Brodie, 2000; Glauser et al., 2013a).
Randomized controlled trials coming of age
This period saw important advances in the application of the randomized controlled trial for the assessment of AEDs. With respect to monotherapy trials, the 1985 study conducted by the Department of Veterans Affairs (VA) collaborative group represents a true milestone. The trial randomized 622 patients with focal and/or focal to bilateral tonic-clonic seizures (partial and/or secondary generalized tonic-clonic seizures according to the terminology used at that time) to receive phenytoin, carbamazepine, phenobarbital or primidone according to a randomized, double-blind, parallel-group design (Mattson et al., 1985). Dosage was adjusted according to clinical response, assisted by serum drug level monitoring, and patients were followed for two years or until treatment failure for lack of efficacy or adverse effects. Overall treatment success was highest with phenytoin and carbamazepine, intermediate with phenobarbital, and lowest with primidone. Carbamazepine, phenytoin and phenobarbital, however, were equally effective in controlling tonic-clonic seizures. As expected, the trial demonstrated clear differences in the adverse effects profile among the various treatments. Interestingly, one of the endpoints used was a composite score which provided a combined measure of seizure control and toxicity, an approach which is being increasingly discussed today to estimate the real clinical impact of different treatments. In subsequent years, the VA collaborative group conducted similarly designed trials which assessed the comparative effectiveness of carbamazepine and valproic acid for the treatment of focal and/or focal to bilateral tonic-clonic seizures (Mattson et al., 1992) and the comparative effectiveness of carbamazepine, lamotrigine and gabapentin in elderly patients with newly diagnosed seizures (Rowan et al., 2005). In evaluating these results, it is often forgotten that the eligibility criteria permitted inclusion of patients already receiving ‘subtherapeutic’ doses or drug levels of AEDs, which were withdrawn during the titration of the study drugs. Because it is now well recognized that many newly diagnosed patients respond at low doses/drug levels (Kwan and Brodie, 2001), the population included in these trials was probably enriched in the proportion of individuals with difficult-to-treat epilepsy. These studies also had a predominantly male population (as many as 93% in the 1992 trial), which could affect to some extent the generalizability of the findings. In any case, the results of the VA trials have been highly valuable, and their influence on the management of epilepsy worldwide remains important to date.
The ‘hyatus’ also saw the establishment of the randomized, double-blind, placebo-controlled adjunctive-therapy trial as the gold standard to evaluate the efficacy and safety of novel treatments for epilepsy. This design was applied to seminal studies that documented the superiority of add-on carbamazepine (Rodin et al., 1974; Kutt et al., 1975) and valproate (Richens and Ahmad, 1975) over placebo in patients with focal seizures. A number of second-generation AEDs entered clinical assessment in the early 80s, and their pivotal studies typically followed a randomized double-blind adjunctive-therapy design. Vigabatrin, in particular, completed European regulatory trials during this period. Interestingly, the sample size in the six double-blind placebo-controlled trials that established the efficacy of vigabatrin (mostly in patients with focal seizures) ranged from 21 to 31 patients only (Mumford and Dam, 1989). One of the reasons for enrolling a small number of patients is that, almost invariably, placebo-controlled trials of AEDs conducted in the 80s used a randomized cross-over design. The advantage of this design is that each individual acts as his or her own control, and thereby a smaller sample size is required compared with the parallel-group design. The cross-over design, however, also has many disadvantages, including a much longer trial duration (which may limit the assessment to one dose only), difficulties in dealing with drop-outs in the analysis of data, and, most important of all, difficulties in excluding or accounting for carry-over effects across study periods (Hills and Armitage, 1979; Sills and Brodie, 2009). For these reasons, this design was soon out of favour with industry and regulatory agencies, and by the early 90s, placebo-controlled adjunctive-therapy trials of AEDs switched to the parallel-group design.
The modern era (1989-2019)
The advent of second-generation AEDs and the new scenarios in drug development
No doubt, a dominating theme of the last 30 years was the introduction of ‘second-generation’ drugs (table 2). The development of these agents was spurred to a large extent by the Anticonvulsant Screening Program, currently known as the Epilepsy Therapy Screening Program, set up in 1975 by J. KIffin Penry at the National Institutes of Neurological Disorders and Stroke (NINDS) of the National Institute of Health (Porter and Kupferberg, 2017). Over the course of its history, the program has tested over 32,000 compounds from more than 600 pharmaceutical firms and other organizations (Kehne et al., 2017), and has played a major role in the development of felbamate, topiramate, lacosamide, retigabine and cannabidiol, and a contributory role in the development of vigabatrin, lamotrigine, oxcarbazepine, and gabapentin (Klein et al., 2017; Porter and Kupferberg, 2017).
When second-generation AEDs started to emerge, it was hoped that the problem of drug-resistant epilepsy could be effectively tackled. Unfortunately, however, only a small proportion of patients with epilepsy resistant to older agents achieve seizure freedom with second-generation AEDs (Gazzola et al., 2007; Prunetti and Perucca, 2011), and seizure outcomes overall have not changed significantly over the last 30 years (Chen et al., 2018). Yet, some (but not all) second-generation AEDs have tolerability advantages over older agents (French and Gazzola, 2011), including a low teratogenic potential (Tomson et al., 2019) and a lower potential to cause enzyme induction and drug interactions (Perucca, 2006). An enlarged pharmacological armamentarium has improved clinicians’ ability to tailor treatment choices to the characteristics of the individual, but also represents a challenge for non-specialists who need to familiarize with the indications, contraindications, optimal dosing schedules, drug interactions, and adverse effects potential of so many drugs.
The currently crowded epilepsy market, the increasing cost of drug development, and the fact that newer compounds with promising findings in seizure models have shown only modest efficacy when tested in patients with drug-resistant epilepsy have discouraged many pharmaceutical companies from investing in AED development. On the other hand, there has been an increasing interest, particularly among small- and medium-size companies, in developing novel molecules for orphan indications where unmet needs are particularly large. In fact, three of the eight AEDs introduced after 2005 (versus none of the 10 AEDs licensed between 1989 and 2005) have been licensed exclusively for the treatment of orphan disorders such as Dravet syndrome (stiripentol, cannabidiol) and Lennox-Gastaut syndrome (rufinamide, cannabidiol) (table 2). More importantly, impressive advances in understanding the molecular causes of epilepsy and mechanisms of epileptogenesis are spurring the development of truly innovative treatments which are no longer targeting the symptoms, but the mechanisms of the disease (Franco et al., 2016). These innovative treatments, some of which might prove to have disease-modifying effects, may include not only novel molecules, but also repurposed drugs currently used for other indications. Targets of special interest in this context include immune-mediated mechanisms and brain inflammation (D’Ambrosio et al., 2013; Ravizza and Vezzani, 2018), and molecular defects resulting from epilepsy gene mutations (Perucca and Perucca, 2019).
Clinical trials in an evolving regulatory scenario
The randomized, double-blind, placebo-controlled adjunctive-therapy trial continues to be the primary tool to obtain regulatory approval of novel AEDs. Implementation of these trials, however, has become increasingly challenging, due to:
- –reluctance of patients to accept prolonged exposure to placebo or a potentially ineffective drug when there are many other approved AEDs on the market that the same patients may have not yet tried (Perucca, 2012);
- –ethical concerns stemming from the finding that exposure to placebo in these trials entails a seven-fold increase in risk of sudden unexpected death (SUDEP) compared with exposure to efficacious doses (Ryvlin et al., 2011);
- –an increase in placebo-associated response over the years, together with a steady reduction in effect size of active treatments (Rheims et al., 2011).
Despite evidence that the decreasing effect size relates to inclusion of less experienced investigators (Perucca, 2012), companies have tried to address it by increasing sample size, which by necessity involves co-opting new and less experienced study sites. To illustrate the magnitude of this trend, pivotal adjunctive-therapy trials of vigabatrin for focal seizures conducted in the 80s were all single-centre studies and enrolled a maximum of 31 patients (Mumford and Dam, 1989), whereas the latest pivotal trial of brivaracetam in patients with the same seizure type enrolled 768 patients in 147 centres, 27 countries, and four continents (Klein et al., 2015). One way to facilitate enrolment in adjunctive-therapy trials, and to address the ethical concerns related to SUDEP risk, is to adopt trial designs that minimize placebo exposure. A feasible option is the time-to-event design, where patients exit the trial after experiencing a predefined number of seizures (for example, pre-randomization monthly seizure count) (French et al., 2015). The advantage of this design, which uses time to exit as primary endpoint, is that patients whose seizures are not improved terminate the trial early. A disadvantage is that duration of exposure varies between the effective treatment arm and the placebo arm, complicating assessment of longer-term seizure response and tolerability. Interestingly, the latest guidelines of the European Medicines Agency (EMA) for the clinical investigation of AEDs, currently in draft form, explicitly state that ‘a time-to-event approach (e.g. time to pre-randomisation monthly seizure count) may be considered’ (European Medicines Agency, 2018). The same guidelines acknowledge its drawbacks, and do not recommend it ‘as the sole study design in the clinical development plan’. Based on this, one option could be to retain the traditional design for the primary trial(s), preferably excluding patients at higher risk for SUDEP, and to apply the time-to-event approach to trials aimed at providing supportive evidence or aimed at extending the indication to other seizure types.
Regulatory requirements to obtain monotherapy indications have traditionally shown a transatlantic divide. The US Food and Drug Administration (FDA) has required demonstration of superiority over a comparator, whereas EMA guidelines require demonstration that the investigational AED is at least as effective as an already established treatment used at optimized doses in patients with newly diagnosed epilepsy followed for at least one year (Perucca, 2008, 2018). FDA requirements have been traditionally met by a variety of trial designs which have in common a comparison with a suboptimal control (often referred to as ‘pseudoplacebo’), but over the years this approach faced increasing ethical concerns. Eventually, a novel approach was adopted whereby seizure outcomes with the investigational treatment were compared with those from historical controls, but this approach also met with criticism (Perucca, 2010). In the last few years, the argument has been repeatedly made that demonstration of efficacy of an AED under adjunctive-therapy use provides sufficient evidence that the same AED will be efficacious also in the monotherapy setting, provided that reasonable steps are taken to exclude efficacy-enhancing interactions with concomitant medications (Mintzer et al., 2015). This argument was recently accepted by the FDA, with perampanel and brivaracetam being the first AEDs to be granted a monotherapy license for the treatment of focal seizures based on data from adjunctive-therapy trials (Perucca, 2018). Interestingly, cannabidiol also received FDA approval for use as mono- and add-on therapy for seizures associated with Lennox-Gastaut syndrome and Dravet syndrome based on data obtained exclusively from adjunctive-therapy trials (Greenwhich Biosciences, 2018). The possibility of extrapolation from the add-on to the monotherapy setting is also mentioned in the latest EMA draft guidelines, which state that ‘on a case by case basis, it may be justified that a monotherapy trial is not necessary to support a monotherapy indication’ (European Medicines Agency, 2018).
The principle of extrapolation is also accepted by regulatory authorities to extend the indication of an AED from adults to children, at least for focal seizures, based on the concept of similarity of disease (Pellock et al., 2017; Perucca, 2018). Of course, when extrapolation of an indication is made from adults to children, the recommended dose regimen takes into account age-related pharmacokinetic differences.
From clinical trials to treatment
A limitation of AED trials conducted in the last decades is that most trials were designed to address regulatory requirements, and did not address issues which are most relevant for clinical practice. For example, the primary question faced by clinicians when using a newly marketed AED is not whether that medication is superior to a placebo, but how it compares with already available drugs, and what is its optimal mode of use. This information is rarely provided by regulatory trials (Perucca, 2018), and the results of many post-marketing comparative trials have been biased by specificities in study protocol that favoured the sponsor's product (Perucca and Wiebe, 2016). A number of academic randomized trials, such as the VA trials (Mattson et al., 1985, 1992; Rowan et al., 2005), the more recent trial of lamotrigine, ethosuximide and valproate for childhood absence epilepsy (Glauser et al., 2013b), and the unblinded Multicentre Study of Early Epilepsy and Single Seizures (MESS) (Marson et al., 2005) did provide highly valuable and clinically relevant information. Yet, as pointed out in an ILAE systematic review of initial treatments for seizure disorders, ‘there continues to be an alarming lack of well-designed epilepsy randomized controlled trials, especially for generalized seizures/epilepsies and in children’ (Glauser et al., 2013a).
Conclusions
Since the landmark studies that led to the discovery of phenytoin, there has been a steady improvement in quality of AED trials, even though most of these trials have been limited in scope to fulfilling regulatory requirements. Knowledge about comparative pharmacokinetics, efficacy, adverse effect profile, and drug interactions of AEDs has improved in parallel, but the quality of evidence guiding treatment choices for most epilepsy syndromes remains suboptimal. Of the large number of drugs that have been introduced for the treatment of epilepsy over the years, many have proven to be highly valuable and provide unprecedented opportunities to tailor drug selection to the characteristics of the individual. Unfortunately, however, newer AEDs have not been found to be more effective than older agents for difficult-to-treat epilepsies, and a change in paradigms for drug discovery and development is needed. Ongoing efforts are aimed at developing truly novel agents which target the underlying causes and mechanisms of epilepsy rather than its symptoms. Testing potential antiepileptogenic/disease modifying medicines will require innovative trial designs and endpoints, and hopefully will lead to introduction of safer and more effective therapies in the future.
Acknowledgements and disclosures
This article was not supported by any funding source. The author received speaker's or consultancy fees from Amicus Therapeutics, Biogen, Eisai, GW Pharma, Sanofi, Sun Pharma, Takeda, UCB Pharma and Xenon Pharma.
* To celebrate the 110th anniversary of the International League Against Epilepsy, Epileptic Disorders is publishing a series of educational manuscripts on ground-breaking topics that have significantly influenced the field of epilepsy, written by past Presidents of the ILAE.