Sang Thrombose Vaisseaux
MENUCollagénopathies COL4A1 et COL4A2 Volume 31, numéro 4, Juillet-Août 2019

Le collagène de type IV est un composant essentiel des membranes basales de l’organisme. Il en existe différents types caractérisés par l’association de trois chaînes (parmi α1 à α6) s’organisant en une triple hélice. La cohésion de chaque hétérotrimère ainsi constitué est assurée par des résidus glycines. Les hétérotrimères constitués de deux sous-unités α1 et d’une sous-unité α2 sont prépondérants et ubiquitaires. Les gènes codant pour les sous-unités α1 (COL4A1) et α2 (COL4A2) sont situés sur le chromosome 13. Les mutations les plus classiquement retrouvées au sein de ces gènes sont situées au niveau des séquences codant pour des résidus glycines et déstabilisent la triple hélice, fragilisant ainsi les membranes basales notamment au niveau vasculaire. La transmission de ces mutations est autosomique dominante. Affectant la même protéine, les mutations des gènes COL4A1 et COL4A2 ont des spectres cliniques très proches. Divers organes peuvent être affectés. Les complications cérébrales, ophtalmologiques et rénales sont les mieux connues. L’âge de début est également très variable : de la vie in utero à l’âge adulte.
Les complications neurologiques
La micro-angiopathie cérébrale
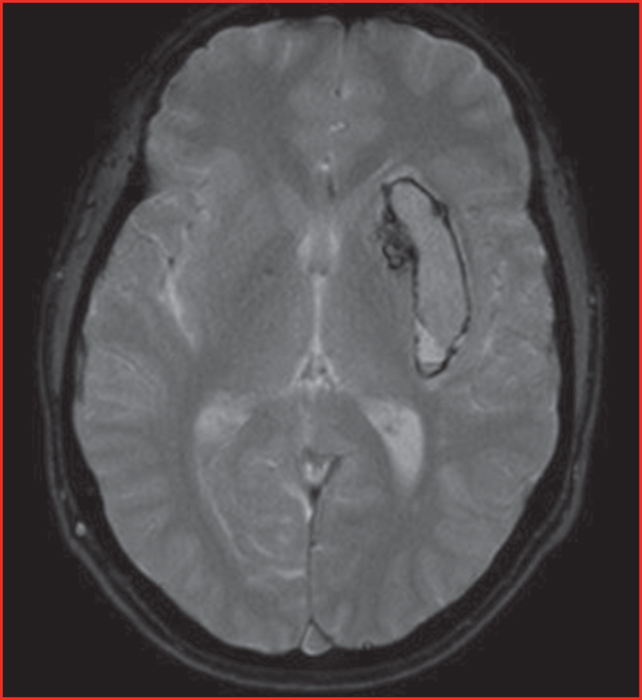
L’hémorragie cérébrale est le plus souvent révélatrice de l’affection (figure 1). Il s’agit dans la plupart des cas d’hémorragies sus-tentorielles profondes, parfois sous-tentorielles ou sous-arachnoïdiennes. Elles peuvent être d’emblée multiples ou récidivantes et survenir à tout âge y compris in utero. Elles peuvent être favorisées par un traumatisme, le passage de la filière génitale de la mère lors de la naissance, la prise d’antithrombotiques (surtout anticoagulants), des efforts physiques intenses, des poussées hypertensives, des épisodes infectieux, etc. [1-3]. A minima, des micro-saignements peuvent être retrouvés sur la séquence T2* à l’imagerie par résonance magnétique (IRM) cérébrale.
Plus rarement, des infarctus cérébraux et des accidents ischémiques transitoires sont décrits. Il s’agirait le plus souvent de petits infarctus lacunaires [4].
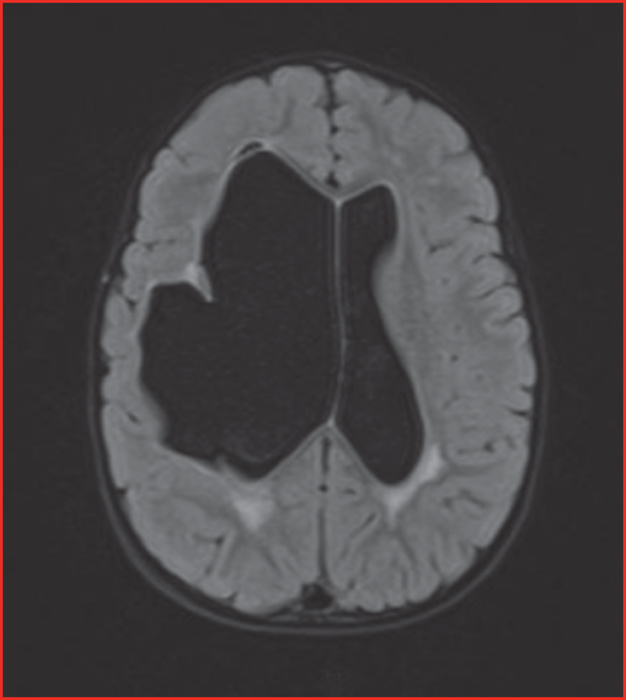
Chez le fœtus ou le jeune enfant, les lésions hémorragiques et ischémiques existent également, parfois qualifiées de lésions « ischémo-hémorragiques ». Leur siège est le plus souvent périventriculaire pouvant conduire à la formation d’une cavité porencéphalique (figure 2) communiquant avec le ventricule. Chez l’enfant, ces lésions peuvent être à l’origine d’un retard psychomoteur, d’une épilepsie parfois pharmaco-résistante, ou d’une hémiplégie infantile. L’apparition de l’hémiplégie infantile pouvant être retardée de plusieurs mois chez le nourrisson, il est souvent difficile de dater l’apparition des lésions à l’imagerie. Des calcifications intracérébrales sont aussi décrites dans ce contexte. Des anomalies de gyration (dysplasies corticales, schizencéphalies) peuvent être observées et pourraient être dues à la survenue de lésions vasculaires au cours du développement cérébral ou à des anomalies de migration des neurones [5]. Des retards de croissance in utero sont aussi décrits chez les enfants mutés. Dans les cas d’emblée sévères avec atteinte encéphalique de mauvais pronostic dépistés lors du suivi échographique systématique de la grossesse, une interruption médicale de grossesse peut être discutée. Il a été montré chez la souris que la césarienne permettait de prévenir les hémorragies périnatales chez le souriceau muté [1]. Par analogie, chez l’homme, il est suggéré de réaliser une césarienne pour l’accouchement des enfants mutés. En revanche, le caractère éventuellement protecteur chez la mère mutée afin de lui éviter des efforts de poussée n’est pas établi.
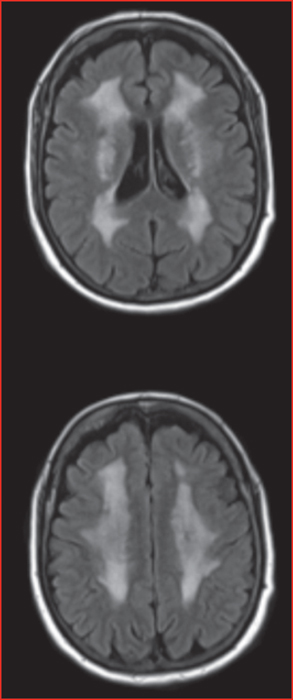
L’atteinte de la substance blanche (figure 3) est très variable pouvant être très étendue, confluente, symétrique, à la fois sus- et sous-tentorielle, épargnant les fibres en U ou limitée à de rares hypersignaux punctiformes de la substance blanche. Elle peut être très marquée chez des sujets asymptomatiques comme être minime, voire absente, chez des sujets ayant déjà fait une hémorragie cérébrale. L’absence de leuco-encéphalopathie ne doit donc pas faire écarter le diagnostic chez un sujet ayant présenté une hémorragie cérébrale.
La macro-angiopathie cérébrale
Certains patients sont porteurs d’anévrismes intracrâniens parfois multiples, le plus souvent situés au niveau de terminaisons et siphons carotides. Bien que leur histoire naturelle ne soit pas connue, leur prise en charge doit être identique à celle adoptée pour les sujets non mutés. A minima, il peut s’agir de dolicho-ectasies artérielles [6].
Une tortuosité des artères cérébrales a été décrite chez certains patients. D’autres artères périphériques extra-cérébrales pourraient également être affectées.
Autres manifestations neurologiques
Des crampes musculaires avec augmentation de la créatine phosphokinase (CPK) et des migraines avec aura ont été décrites dans certaines familles.
Les signes ophtalmologiques
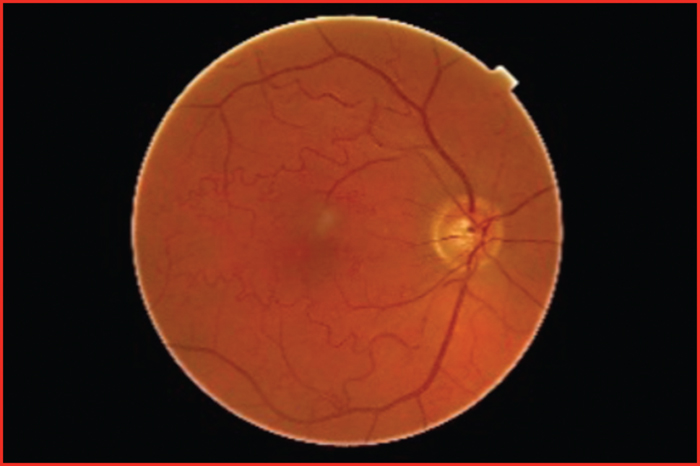
Les tortuosités artériolaires rétiniennes sont très évocatrices des mutations COL4A1 ou COL4A2 (figure 4) mais sont inconstantes et peuvent apparaître au cours de la vie. Elles peuvent se compliquer d’hémorragies rétiniennes responsables de pertes visuelles ou être asymptomatiques, découvertes uniquement à l’examen du fond d’œil. L’angiographie rétinienne n’est pas indispensable au diagnostic. Les collagénopathies COL4A1/A2 exposent au risque d’hémorragies oculaires, de décollement de la rétine, et de cataractes juvéniles parfois favorisées pas des traumatismes même mineurs. D’autres anomalies malformatives sont rapportées comme les dysgénésies du segment antérieur de l’œil [4] (syndrome d’Axenfeld-Rieger), les anomalies du nerf optique, la microphtalmie, etc.
Les anomalies extra-neuro-ophtalmologiques
L’atteinte rénale liée à des mutations COL4A1 se manifeste de façon variable par une hématurie macroscopique ou microscopique, une insuffisance rénale et des kystes rénaux. Le parenchyme rénal a pu être étudié histologiquement montrant des irrégularités et des épaississements focaux des membranes basales tubulaires et de la capsule de Bowman sans anomalie de structure de la membrane basale glomérulaire. L’atteinte rénale est particulièrement importante dans le phénotype HANAC (Hereditary Angiopathy, Nephropathy, Aneurysms, and muscle Cramps), lié à certains types de mutations, pouvant s’associer à des troubles du rythme cardiaque supraventriculaire (fibrillation auriculaire), un syndrome de Raynaud, des kystes hépatiques et un prolapsus de la valve mitral [7]. Même s’il n’est pas nul, le risque hémorragique cérébral pourrait être moindre avec ce phénotype.
Les porteurs asymptomatiques
L’étude systématique des familles a permis de mettre en évidence des sujets mutés asymptomatiques sur le plan clinique voire paraclinique. L’expression phénotypique est très variable d’un sujet à l’autre d’une même famille. Des mutations de novo en mosaïque à un stade plus tardif du développement embryonnaire pourraient expliquer chez certains une atteinte clinique moins sévère et le fait que certains organes soient épargnés. Par ailleurs, la pénétrance incomplète pourrait être expliquée par l’influence de facteurs externes environnementaux (traumatiques) ou de facteurs internes (interaction entre gènes, facteurs de risque cardiovasculaires associés) dans la genèse des complications notamment vasculaires. Ces observations rendent le conseil génétique délicat [8].
Diagnostic et intérêt
Le diagnostic doit être évoqué dans le bilan d’une hémorragie cérébrale du sujet jeune sans cause retrouvée après un bilan complet, compte tenu des conséquences possibles sur la descendance du patient. Un diagnostic anténatal peut être proposé dans certains cas. Un accouchement par césarienne est systématiquement proposé pour la naissance des enfants mutés.
La suspicion diagnostique peut être étayée par la recherche d’antécédents personnels et familiaux d’accidents vasculaires cérébraux, d’anévrismes intracrâniens, d’hémiplégies infantiles, d’hémorragies ou de malformations oculaires, de cataractes juvéniles et de maladies rénales, sans oublier la recherche d’antécédents obstétricaux, de fausses couches notamment tardives, d’interruptions médicales de grossesse, de morts fœtales.
La réalisation d’un fond d’œil est utile car si les anomalies sont inconstantes, la découverte de tortuosités artériolaires rétiniennes par un ophtalmologue expérimenté est fortement évocatrice du diagnostic. Le diagnostic de certitude est génétique par la recherche de mutation de ces gènes dans le sang1. Le dépistage des apparentés asymptomatiques doit répondre à des règles de consultation pluridisciplinaire permettant une concertation entre clinicien, psychologue, et généticien laissant un délai de réflexion puisqu’il s’agit d’affections pour lesquelles aucun traitement n’est disponible. Cependant, la découverte d’anévrismes intracrâniens à l’imagerie cérébrale peut parfois déboucher sur un traitement endovasculaire voire neurochirurgical à visée préventive avant une éventuelle rupture. Dans la plupart des cas, les anévrismes découverts fortuitement ne justifieront pas d’un traitement et une simple surveillance régulière sera envisagée. Il faut donc prendre en compte le retentissement psychologique potentiel, notamment chez les sujets asymptomatiques, de se savoir porteurs d’anévrismes intracrâniens.
Sur le plan préventif, il est recommandé aux sujets mutés d’éviter certaines activités à risque de traumatisme, les activités physiques intenses et la prise d’antithrombotiques. La prise de ces traitements doit être limitée à leurs indications formelles en réévaluant régulièrement la balance bénéfice/risque. Le risque chirurgical n’est pas connu et dépend probablement du type de chirurgie.
Bilan initial et suivi
En cas d’identification sur les gènes COL4A1 ou COL4A2, un bilan général et systémique est proposé en première intention :
- –IRM cérébrale avec des séquences de diffusion, T2-FLAIR, T2*, 3DTOF du polygone de Willis ;
- –échographies abdominale et rénale ;
- –examens biologiques : dosage de la créatininémie, CPK, recherche de protéinurie et d’hématurie ;
- –électrocardiogramme ;
- –consultation d’ophtalmologie avec fond d’œil ;
- –consultation de neurologie ;
- –consultation spécialisée selon les organes atteints (consultation de néphrologie, médecine vasculaire, etc.) ;
- –consultation avec un généticien (conseil génétique).
Le rythme de surveillance est à adapter aux manifestations cliniques mais le suivi doit être régulier. Des procédures et recommandations de suivi devraient être mises en place prochainement.
Autre type de mutation affectant le gène COL4A1 : PADMAL
PADMAL (Pontine Autosomal Dominant MicroAngiopathy with Leukoencephalopathy) a été décrit dans des familles présentant des infarctus cérébraux récidivants dès l’âge de 35-45 ans et prédominants au niveau pontique. Ils sont responsables d’un déficit moteur et cognitif d’aggravation progressive ou par à-coups. Récemment, il a été montré que ce syndrome était lié à une mutation au niveau d’une région non codante du gène COL4A1 correspondant à un site d’accrochage d’une protéine régulant l’expression du gène. Ce type de mutation est responsable d’une surexpression du gène COL4A1[9].
Conclusion
Les mutations des gènes COL4A1 et COL4A2 sont une cause rare de micro-angiopathie cérébrale et d’hémorragie cérébrale quel que soit l’âge. De pénétrance incomplète, leur expression phénotypique est très variable au sein d’une même famille et leur spectre clinique est très large et encore mal connu, nécessitant un bilan systémique complet. Il faut savoir les évoquer notamment chez les sujets en âge de procréer compte tenu du risque pour l’enfant de développer précocement la maladie.
Liens d’intérêts
l’auteur déclare ne pas avoir de lien d’intérêt.
1 Informations disponibles sur www.cervco.fr
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International