Hématologie
MENULes lymphomes anaplasiques à grandes cellules associés aux prothèses mammaires : du diagnostic au séquençage moléculaire Article à paraître

Breast implant-associated anaplastic large cell lymphoma (BI-ALCL) lacking expression of anaplastic lymphoma kinase (ALK-) was first described in 1997 by Keech and Creech [1]. It is a provisional entity that was recently recognised in the updated 2017 classification by the World Health Organization (WHO) [2]. It is similar to the other subtypes of ALCL, including systemic ALK- and ALK+, and primary cutaneous ALCL, and shares morphological and phenotypic characteristics with these subtypes based on tumour proliferation of large cells, often with a reniform nucleus, and constant expression of CD30[2].
BI-ALCL remains rare, representing 0.06% of all non-cutaneous lymphomas registered in the national lymphoma review network (Lymphopath) funded by The Institut National du Cancer (INCa) [3-5]. However, since its initial description in 1997, the number of cases of BI-ALCL has constantly increased with, to date, more than 70 patients affected in France. This growing incidence prompted national health organisations (INCa, ANSM, the Directorate General for Health [Direction Général de la Santé, DGS]) to step up monitoring and surveillance of patients with breast implants. Recently, numerous studies, particularly molecular studies, have made it possible to better characterise the biology of this new subtype. New investigations remain to be undertaken in order to better understand the pathogenesis of this disease and to identify all the genetic or environmental risk factors which will eventually make it possible to limit and prevent the development of BI-ALCL in women with implants [6].
Epidemiology
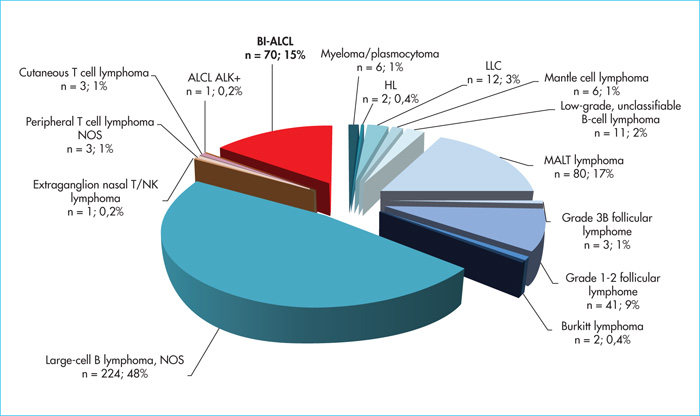
Primary breast lymphomas account for 1–2% of all non-Hodgkin's lymphomas and less than 0.5% of breast cancers. The incidence of BI-ALCL remains low, ranging from 0.1 to 5 cases per 100,000 women with breast implants depending on the series [7-10]. The first case was described by Keech and Creech in 1997 [1] who reported a case of BI-ALCL occurring in a periprosthetic effusion around a saline-filled breast implant. In 2008, de Jong et al.[11] reported an association between the occurrence of an ALCL and breast implant surgery, leading the Food and Drug Administration to issue an alert in 2011 [12]. Since then, nearly 600 cases have been recorded worldwide. In the Lymphopath cohort [3], representing the largest prospective cohort of lymphoma patients to date, with more than 60,000 primary lymphoma diagnoses recorded since 2010, 468 breast lymphomas have been diagnosed (figure 1). Of these, B-cell lymphomas are by far the most common (n = 392), with a majority of diffuse large B-cell lymphomas (DLBCL) (n = 224, 48%), followed by extranodal marginal zone B-cell lymphomas of mucosa-associated lymphoid tissue (MALT) lymphomas (MALT lymphoma) (n = 80, 17%) and Grade 1–2 follicular lymphomas (n = 41, 9%). T cell lymphomas represent only 16% (n= 78) of primary breast lymphomas but with a large majority of ALK-ALCL (n = 70; 15% of breast lymphomas and 90% of PCTL), all associated with breast implants. While some initially suggested that this association might be fortuitous, the increasing number of cases reported in the literature now suggests the contrary [4, 7-11]. According to de Boer et al.[9] the relative risk of developing BI-ALCL in women with breast implants was 421.8 (odds ratio) with the probability of occurrence (absolute risk) increasing with age from 29 per million at age 50 to 82 per million at age 70. This risk of occurrence appears to be higher in patients with textured breast implants than in those with smooth implants [4, 9, 10, 13]. This has also been observed in our French series in which almost all BI-ALCL patients had textured prostheses [4, 13], most often Biocell Allergan implants, which were withdrawn from the market in France and around the world in 2019.
Clinical presentation
The average age at diagnosis of BI-ALCL is 61 years old (ranging from 28 to 87 years old). By definition, all BI-ALCL patients underwent a breast implant surgery for cosmetic or reconstructive purpose.
In the majority of cases, the breast implants associated with BI-ALCL are textured and most often filled with silicone gel [4, 13]. The median time interval from implant placement to diagnosis of BI-ALCL is approximately 8–10 years, ranging from 0.2 to 30 years [4-6, 13], however, it is sometimes difficult to determine this formally as some BI-ALCL patients have been re-implanted several times, sometimes with different types of implants. This was the case, for example, in our French series, and also explains why it is sometimes difficult to incriminate a specific type of implant [5, 13].
BI-ALCL may present two distinct clinical forms at the time of diagnosis [4, 13, 14]. Most patients (80%) present with a periprosthetic effusion, known as a peri-implant seroma with no tumour mass and no remote extension; these patients have an excellent prognosis. A minority of patients (20%) present a mammary tumour mass which can spread outside the breast and has a poorer prognosis [4, 13, 14].
Histopathological diagnosis of breast implant-associated large-cell anaplastic lymphoma
The diagnosis of BI-ALCL is carried out by cytological analysis of a periprosthetic effusion and/or histological analysis of capsule fragments. Reaching this diagnosis is sometimes difficult and requires adequate sampling of the capsule and complementary immunohistochemical (IHC) or molecular examinations. In France, each BI-ALCL diagnosis must be reported to the ANSM via Lymphopath, the INCa national network for lymphoma expert review [3].
Morphologically, there are two histological subtypes [4].
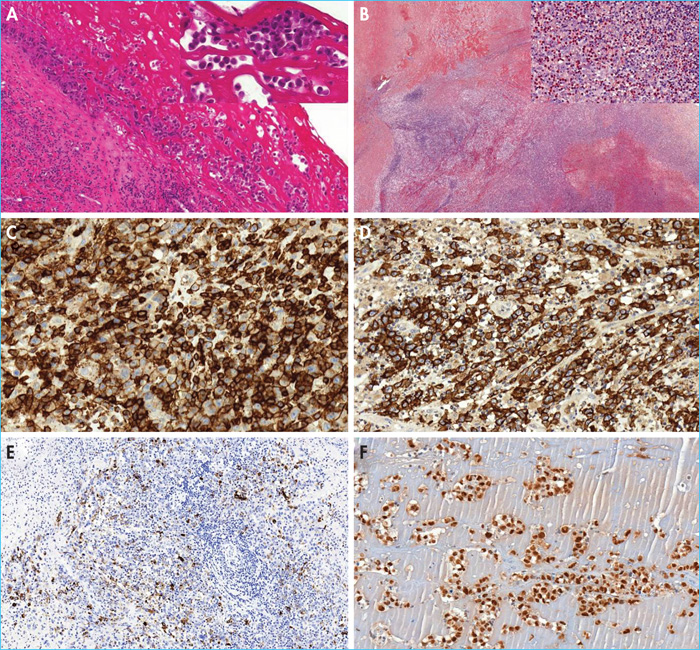
- –The in situ subtype; the most common subtype observed in patients diagnosed with isolated periprosthetic effusion. Samples from prosthetic capsules are infiltrated by chronic inflammatory fibrosis with lymphoplasmacytic cells and/or reactive histiocytes. The inner surface of the capsule is lined by proliferating tumour cells that forms single or multi-cellular layers, often mixed with serofibrinous material (figure 2). The cytological aspect reveals medium- to large-sized tumour cells, with an atypical nucleus which is often hyperchromatic, ovoid or curved, sometimes reniform or horseshoe-shaped, providing the cytological characteristics of hallmark cells.
- –The “infiltrative” subtype; observed in patients presenting with a breast mass at diagnosis. This subtype represents a proliferating tumour that infiltrates the capsule, is poorly limited, and sometimes extends to the breast parenchyma or pectoral muscle. This tumour proliferation consists of large cells with a strongly nucleated mono- or bi-nucleated nucleus resembling Hodgkin's or Reed-Sternberg cells, which are sometimes numerous(figure 2). These tumour cells are often accompanied by necrosis and sometimes sclerosis with a reactive inflammatory population including histiocytes, lymphocytes, plasma cells and many eosinophils.
More rarely described, the “mixed” subtype combines the two morphological aspects, suggesting a continuum between the two histological subtypes.
Regardless of the histological subtype, the tumour cells are strongly and uniformly positive for CD30(figure 2). Neoplastic cells are also often positive for EMA. ALK is always negative. Almost consistently, BI-ALCL has an incomplete T phenotype with loss of one or more T cell markers (CD3, CD2, CD5, CD7) in the majority of cases, with the exception of CD4 (figure 2), which is generally present (figure 2). Tumour cells have an activated cytotoxic profile with expression of granzyme B and/or perforin in addition to TiA1(figure 2). Anti-CD43 and CD45 antibodies are almost consistently positive. In situ hybridisation for Epstein-Barr virus (EBV) is negative. Finally, the tumour cells are predominantly positive for anti-phospho-signaltransducer and activator of transcription 3 antibody (p-STAT3), illustrating the constitutive activation of STAT3 in this pathology (figure 2) [4].
Molecular and genetic characteristics
Studies on the rearrangement of the T-cell receptor genes γ and/or β most often demonstrate a clonal rearrangement. This rearrangement is sometimes absent or oligoclonal, probably related to the low proportion of tumour cells present in the sample, especially in in situ BI-ALCLs.
Unlike ALK+ ALCLs, BI-ALCLs do not exhibit an ALK rearrangement. They also do not show any rearrangement of the DUSP22-IRF4 or TP63 genes, which are present in 20% and 8% of ALK- ALCLs, respectively [2].
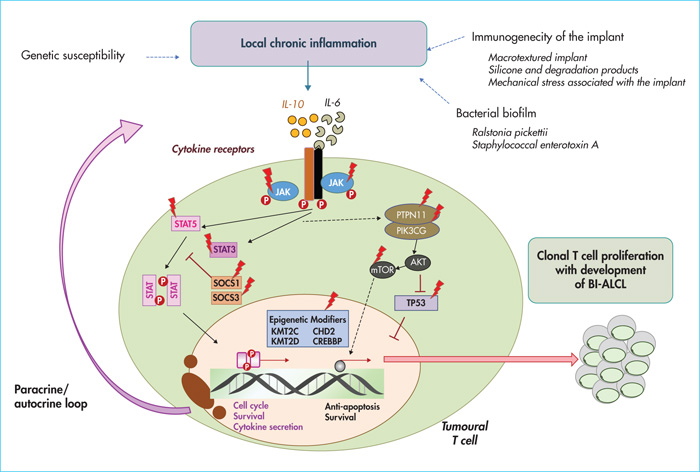
Nevertheless, like ALCLs, BI-ALCLs activate the Janus Kinase (JAK)/STAT pathway, that could be due to the presence of recurrent mutations in the STAT3 gene, initially observed in 26-64% of cases based on high-throughput sequencing of small series or isolated cases, and could be also linked to the cytokine environment of BI-ALCLs rich in IL-6 and IL-10, secreted by tumour cells in vitro (for a review, see reference [5]).
More recently, whole-exome sequencing of the largest BI-ALCL series has enabled the following findings:
- –The frequency of gene mutations involved in the JAK/STAT pathway (59% of cases), which are more frequent in the infiltrating subtype than in the in situ subtype, and which include activating mutations of STAT3 (38%), JAK1 (18%) and STAT5B (3%), but also mutations in the negative regulators of the JAK/STAT pathway such as SOCS3 (6%), SOCS1 (3%) and PTPN1 (3%) (figure 3) [15].
- –Identification of a preponderance of molecular alterations in epigenetic regulators observed in 74% of cases with, in particular, somatic mutations in KMT2C (26%), KMT2D (9%), CHD2 (15%) and CREBBP (15%) genes. Other mutations affecting genes involved in lymphocyte differentiation (12%) such as EOMES or in the phosphatidylinositol-kinase type 3 (PI3K)-AKT/mTOR pathway (6%), as well as TP53 loss of function mutations (12%), have also been identified. Finally, in this study, recurrent cytogenetic abnormalities such as gains on chromosomes 2, 9p, 12p and 21 and deletions on 4q, 8p, 15, 16 and 20 were observed [15]. Further analyses of copy number variations in one isolated case showed gains associated with 19p and loss of chromosome at 10p and 1p (for review: [5]).
Comparative analysis of gene expression between BI-ALCL and cutaneous ALCL initially showed similarities between lines derived from BI-ALCL patients and cutaneous ALCL, with a comparable TH17/TH1 cytokine profile and over-expression of the transcription factors SOCS3, SATB1 and JunB. More recently, transcriptomic analysis of samples from patients with BI-ALCL showed that BI-ALCL and ALCL have a distinct gene expression profile with the exception of the common activation of STAT3 and inhibition of signalling induced by TCRs [16, 17]. Indeed, BI-ALCL may be distinguished from ALCLs by over-expression of genes involved in hypoxia (carbonic anhydrase CA9, VEGFA, VEGFB, SLC2A3) [16], cell mobility (CCR6, MET and HGF), myeloid differentiation (PPARG and JAK2) and viral transcription (RPS10) [17].
Differential diagnosis
Differential diagnoses include the other CD30+ T lymphoproliferative disorders.
Systemic anaplastic large-cell ALK+ lymphoma
This is a tumour of large cells with horseshoe-shaped nucleus, referred to as hallmark cells. There are several subtypes:
- –classic subtype (hallmark cells with marked sinus infiltration);
- –small-cell subtype;
- –lymphohistiocytic subtype;
- –Hodgkin-like subtype;
- –Mixed subtype.
Immunohistochemically, tumour cells are ALK+ by definition, a consequence of the rearrangement of the ALK gene, and also express CD30 and EMA. The cells are most often negative for T markers, except for CD4 or CD43. Proteins associated with cytotoxic granules (granzyme, perforin and TIA1) are frequently expressed. Finally, at the gene level, a rearrangement of the ALK gene is constant, the most frequent partner of which is the gene encoding nucleophosmin (NPM), secondary to t(2;5) translocation, providing nucleocytoplasmic expression of ALK [2].
Systemic ALK- anaplastic large-cell lymphoma
This is a tumour with a morphology comparable to that of conventional ALK+ ALCLs. Phenotypically, the cells are ALK- and CD30+ and EMA+, sometimes CD3+ or positive for other T cell markers, and express proteins associated with cytotoxic granules. At the gene level, in 20% of cases, a mutation in JAK1 or STAT3 is observed, leading to activation of the JAK/STAT pathway. Recently, two subgroups of ALCL ALK- patients with significantly different prognoses have been described: ALCL ALK- patients with DUSP22-IRF4 (locus 6p25) rearrangement with a likely good prognosis (25% of cases) and those with rearrangement in TP63 (8% of cases) which seem to be associated with a worse prognosis [2]. Fusions involving the ROS or TYK genes, which can lead to activation of STAT3, have also been reported [18].
Cutaneous T CD30+ lymphoproliferative disorders
Primarily cutaneous CD30+ lymphoproliferative disorders consist mainly of primary cutaneous ALCL (c-ALCL) and lymphomatoid papulosis (LP). Between these two extremes of the spectrum of cutaneous CD30+ T lymphoproliferation, there are borderline forms which are difficult to classify. Clinically, c-ALCL is most often characterised by a single large nodule or a few nodules localised to an area, often ulcerated, and may regress but never completely. Histologically, c-ALCL consists of an infiltrate of large CD30+ anaplastic cells in the dermis and sometimes the hypodermis. The tumour cells are CD3+, CD4+, CD8- (sometimes CD8+), EMA- and ALK-. At the gene level, DUSP22-IRF4 rearrangements are observed in 25% of cases. Other gene abnormalities have been described, such as NPM1-TYK2 rearrangement leading to activation of the JAK/STAT pathway [2]. On the other hand, the presence of TP63 gene rearrangement in c-ALCL remains anecdotal.
LP is a chronic and recurrent skin eruption of small papules at different ages, sometimes ulcerated, and can be more or less generalised. The histological characteristics of LP are variable. Several histopathological forms can be distinguished, the most frequent of which are:
- –LP type A (conventional LP), characterised by a triangular appearance of the lesion, containing large anaplastic cells accompanied by a histiocytoid polymorphic infiltrate;
- –LP type B (mycosis fungoïdes-like);
- –LP type C (c-ALCL-like);
- –LP type D (cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma-like);
- –LP type E (angiocentric);
- –LP type F (follicular).
These cells are CD30+, CD3+ and CD4+, more rarely CD8+ (mainly in LP type D). Genetically, DUSP22 rearrangements are less frequently found than in c-ALCL.
Clinically, LP and c-ALCL are characterised by an excellent prognosis and a less aggressive, wait-and-watch treatment.
Apart from LP and c-ALCL, CD30+ transformed mycosis fungoides represents another CD30+ T lymphoproliferative disorder with a worse prognosis.
Other CD30+ lymphomas
Classic Hodgkin's lymphoma is characterised by the presence of CD30+ Hodgkin's or Reed-Sternberg cells, CD15+, losing expression of pan B-markers CD20 and CD79a and weakly expressing PAX5.
Extranodal natural killer (NK)/T-cell lymphoma, nasal type is a lymphomatous proliferation with NK phenotype (most often) CD3ε+, CD56+, and CD30+, expressing cytotoxic molecules, generally CD8- or more rarely a cytotoxic T cell phenotype expressing CD8 and TCRγδ or αβ. The tumour cells are EBER+ [2].
TNM stage, treatment and prognosis
Patient management and the prognosis of this disease are closely linked to the clinical presentation of BI-ALCL at diagnosis (seroma versus tumour mass) and the degree of extension of the tumour, evaluated according to the TNM (tumour node metastasis) staging [12, 19]. Evaluation is achieved by meticulous and complete histological analysis of the periprosthetic capsules which determines local tumour invasion of the BI-ALCL:
- –T1 corresponds to tumour proliferation confined to the periprosthetic effusion and/or lining of the internal surface of the capsule without infiltrating it;
- –T2 corresponds to minimal capsular invasion;
- –T3 corresponds to a clear invasion of the capsule;
- –T4 corresponds to tumour infiltration beyond the capsule, with or without lymph node involvement.
Node (N) and metastatic (M) involvement is assessed by imaging including breast MRI and positron emission tomography (PET-CT). Since 2016, a WebEx national multidisciplinary meeting has been implemented by the French Cancer Agency in order to define therapeutic strategies for new BI-ALCL patients after histological confirmation by the Lymphopath Network. At the same time, each consenting patient could be registered in the BI-ALCL registry funded by LYSA/LYSARC in order to collect ambispectively clinical data, including reasons for breast implantation (breast augmentation, reconstruction), implant manufacturer, treatments and outcome [13]. BI-ALCL patients with in situ subtype are successfully treated with implant removal and capsulectomy. On the other hand, patients with a tumour mass at diagnosis require total capsulectomy combined with adjuvant chemotherapy. Recently, the use of vedotine brentuximab (anti-CD30 antibodies coupled with monomethyl auristatin) in combination with chemotherapy has been recommended, taking into account the demonstrated benefit of the addition of this drug for systemic ALCL. BI-ALCL has an excellent prognosis, with an overall survival at three and five years of 93% and 89%, respectively [19]. In our series, most patients with in situ BI-ALCL related to isolated periprosthetic effusion are considered to be cured and free of disease after five years of median follow-up [4, 13]. Nevertheless, the infiltrative subtype associated with a breast mass appears to have a worse prognosis, with an overall survival of 75% at five years, requiring a more intensive therapeutic approach, with anthracycline-based chemotherapy [4, 13]. In both cases, medium and long-term follow-up is recommended.
Physiopathology
Most studies highlight the potential role of chronic antigenic stimulation in the pathogenesis of BI-ALCL. Indeed, the chronic inflammatory reaction secondary to chronic antigenic stimulation may promote the release of oxygen radicals, micro-RNA instability and epigenetic modifications, leading to genetic instability and supporting the emergence of a tumour clone. The best example illustrating this mechanism is that of MALT lymphoma for which an underlying chronic infection or autoimmune phenomena play a key pathogenic role in the evolution and progression of the tumour. In the context of BI-ALCL, this chronic immune response may be secondary to the immunogenicity of the implant or its filler when it breaks. This latter hypothesis has been highlighted by some studies showing that silicone gel and its degradation products are directly toxic by causing fibrosis and inflammation in tissues, and may help maintain T-cell proliferation (for a review, see reference [5]). However, although initial hypotheses tended to preferentially incriminate the silicone filling gel, this is not solely responsible, since, to date, most of the cases described occurred without prosthetic rupture and others, although rarer, developed on a breast prosthesis filled with physiological saline [4]. More recently, the bacterial biofilm of implants has been incriminated through its contamination from the breast and skin microbiome during surgery. These microorganisms could trigger an immune response, for example, by stimulating toll-like receptors on the surface of immune cells, and induce T cell activation and proliferation that may promote clonal expansion (for a review, see reference [5]). Some recent analyses of bacterial biofilm have suggested a high proportion of Ralstonia pickettii, a common Gram-negative contaminant of drinking water, in samples with BI-ALCL compared to non-tumour capsules, suggesting a pathogenic role of the specific microbiota associated with BI-ALCL [20]. In addition, macrotextured implants (Biocell textured implant), with a higher bacterial load and rougher surface, would be associated with a more marked proliferation of T cells and a greater risk of development of BI-ALCL [21].
The hypothesis of chronic antigenic stimulation is reinforced by the in vitro study by Kadin et al.[22] who showed that a cytokine and transcriptional profile of tumour lines derived from BI-ALCL patients (TLBR1, TBR2 and TLBR3) was comparable to that found in T lymphocytes after bacterial stimulation, in particular, with polarisation of T helper (Th)1/Th17 type T cells associated with a high expression of interleukins (IL)-17 and interferon γ (IFN-γ).
The immune microenvironment participates in vitro in the survival of tumour cells of BI-ALCL lines (TLBR1, TBR2 and TLBR3) via the cytokines IL-2, IL-6 and IL-10 present in the culture medium. On the other hand, autocrine secretion of immunosuppressive cytokines, such as IL-6, transforming growth factor β (TGFβ) and IL-10 by tumour cells, is believed to promote tumour escape by inducing an expansion of protumoural regulatory cells such as T-regulators (T-reg), TAMs (for tumour associated macrophages) and poorly differentiated myeloid cells with suppressive MDSC (myeloid derived suppressive cell) function (for a review, see reference [5]).
Finally, activation of the JAK/STAT signalling pathway – secondary to the response to cytokines IL-6 and IL-10, or directly, via molecular alterations activating the JAK/STAT pathway - would participate in the development of BI-ALCLs [5, 15]. Other transcription factors such as JunB and STATB1 (special AT-rich sequence-binding protein-1), that promote the proliferation of CD30+ T cells over-expressed in BI-ALCL, as well as other recently described mutations involving epigenetic regulators and TP53 could be directly involved in the oncogenesis of BI-ALCL [5, 15].
Ultimately, a set of intrinsic and/or extrinsic factors involving a chronic inflammatory process linked to immunogenicity of the implants or their filler, the microbiota, genetic susceptibility (in particular the HLA A26 antigen) and the acquisition of mutations or molecular alterations may favour clonal expansion of T lymphocytes and lead to the appearance of BI-ALCL (figure 3).
Conclusion
BI-ALCL is a rare disease that was recently reocognised in the new WHO 2017 classification as a new entity of mature T-cell lymphoma that develops selectively on breast implants [2]. Its diagnosis is based on morphological analysis of the capsule or periprosthetic effusion but may require immunohistochemical or even molecular investigation when there is any clinical suspicion of BI-ALCL. BI-ALCL patients may have peri-implant seroma and/or breast mass (aesthetic or as part of reconstruction following a mammectomy for breast cancer). Note that a higher frequency of BI-ALCL is reported for Allergan macrotextured prostheses with Biocell texturing, which were withdrawn from the market based on the decision by the Agence Nationale de Securite des Medicaments. Causal factors are environmental and genetic, involving the host immune system in response to chronic antigenic stimulation (textured implants? bacterial biofilm? genetic predisposition?), as well as activation of the JAK/STAT signalling pathway (secondary to mutations or linked to the cytokine environment) and the frequent alterations in epigenetic regulators. All of these factors may lead to activation and proliferation of cytotoxic T cells which may eventually lead to clonal expansion, favouring the development of BI-ALCL. The prognosis for the disease is generally good with an overall three-year survival rate of more than 90%.
Conflicts of interest
Corinne Haioun and Philippe Gaulard received honoraria from Takeda.