Epileptic Disorders
MENUResponse to lacosamide monotherapy in a patient with medically refractory Jeavons syndrome: a case report and review of the literature Volume 22, numéro 5, October 2020

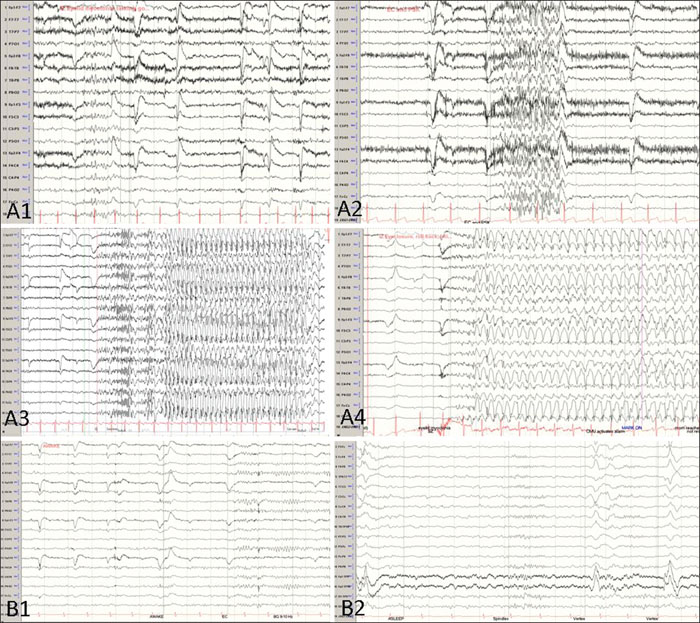
- Mots-clés : Jeavons syndrome, eyelid myoclonia, lacosamide, generalized epilepsy
- DOI : 10.1684/epd.2020.1198
- Page(s) : 643-7
- Année de parution : 2020
Jeavons syndrome is a childhood genetic generalized epilepsy characterized by eyelid myoclonia with or without absences, eyelid closure-induced epileptiform discharges and/or seizures and photoparoxysmal response. This syndrome accounts for up to 12.9% of generalized epilepsies, however, it is frequently under-reported. The utility of lacosamide in genetic generalized epilepsy and Jeavons syndrome is unclear. We present a case of a 15-year-old female with medically refractory Jeavons syndrome with seizure resolution in response to lacosamide monotherapy at standard daily doses. She had failed treatment with adequate trials of ethosuximide, valproic acid, lamotrigine, topiramate and the ketogenic diet, either as monotherapy or in combination. The frequency of seizures was confirmed in the epilepsy monitoring unit. She was treated with a loading dose of 200 mg of intravenous lacosamide and started at a maintenance dose of 100 mg, twice daily. The EEG showed a dramatic response with resolution of seizures and dramatic improvement in interictal discharges. She remained seizure-free for 11 months on lacosamide monotherapy after which seizures recurred in the setting of medication non-compliance. This highlights the potential role of lacosamide as an option in this syndrome if other drugs are ineffective or not tolerated.