Epileptic Disorders
MENUPhotoparoxysmal response in ADCK3 autosomal recessive ataxia: a case report and literature review Volume 23, numéro 1, February 2021

The AarF domain-containing kinase 3, ADCK3 (also named CABC1; a human homologue of the yeast ABC1/COQ8 gene), is an autosomal mitochondrial gene located on 1q42.13 [OMIM#606980], encoding an ancestral regulatory protein belonging to the family of atypical kinases (including phosphoinositide and choline kinases). This kinase plays a regulatory role in indirect ubiquinone biosynthesis, due to demonstrated Mg2+-dependent ATPase activity [1]. Moreover, it has an important role in the biosynthesis of coenzyme Q10 (CoQ10), a lipid-soluble electron transporter, as part of a feedback loop that regulates ATP production and therefore essential for aerobic cellular respiration. The precise biological role of ADCK3 is still unknown [2].
Clinical phenotypes of patients with ADCK3 mutations were first described in association with an atypical form of autosomal recessive cerebellar ataxia (ARCA2) [3, 4]. Thereafter, a broad phenotypic spectrum has been described, including the most typical presentation characterized by progressive ataxic features and cerebellar atrophy, variably combined with spasticity, muscular involvement (hypotonia, exercise intolerance), neurosensory impairment, intellectual disability, seizures, stroke-like episodes (SLE), migraine and psychiatric symptoms [5]. Severity is variable, ranging from early and rapid deterioration to milder adult-onset forms, and genotype-phenotype correlation does not appear to be predictable.
Epilepsy has been reported in ADCK3 patients, more frequently than in other CoQ10 deficiency syndromes, accounting for 20-30% of cases [6, 7]. The most frequent epileptic phenotypes are posterior focal to bilateral tonic-clonic seizures, with a higher risk of status epilepticus and epilepsia partialis continua (EPC). Even though seizures and progression of the disease have been related to clinical and radiological onset of SLE, the mechanism of epileptogenic threshold lowering is unclear, since several cases can present with seizures even in the absence of ischaemic damage [7].
We describe the case of a young girl with infantile onset of ataxia and epilepsy and electroencephalographic evidence of photoparoxysmal response (PPR).
Case study
The subject is a 15-year-old female, the second child of unrelated healthy parents. No neurological or psychiatric family history was documented. Gestation was threatened by abortion in the first trimester. Karyotype following amniocentesis resulted normal. The patient was born at term with no distress at birth; anthropometry was appropriate for age. At examination, a 2/6 cardiac murmur was documented and echocardiogram showed mild pulmonary supravalvular stenosis. Developmental stages were normal for age, except for a mild language delay. Hypermetropic astigmatism was diagnosed in infancy.
At the age of two, because of frequent falls and imbalance, the patient was referred to a paediatric neurologist who documented scoliosis and suggested speech therapy. For the persistence of language difficulties, clumsiness and gait problems, the patient was admitted to our institution at seven years of age. Neurological examination showed dysarthria with scanning speech. Difficulties in chewing and swallowing harder foods were reported. No abnormalities of eye gaze or ocular motility were observed; muscle trophism and tendon reflexes were normal. Intention tremor and dysmetria were present, and autonomous walking was possible with ataxic gait. The Wechsler Preschool and Primary Scale of Intelligence (3rd edition) showed a total Intelligence Quotient (IQ) of 68.
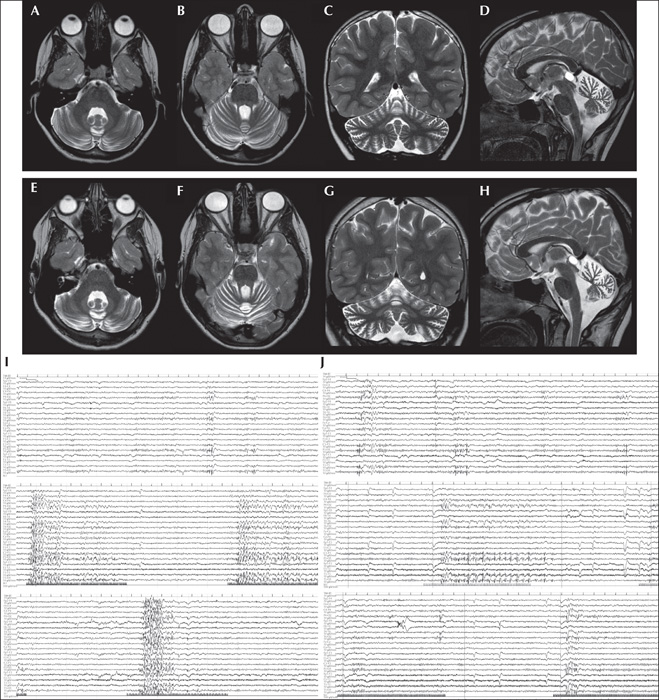
Brain MRI, performed at seven years of age and repeated at 11, showed slowly progressive cerebellar atrophy with prevalent involvement of the vermis (figure 1A-H). Spectroscopy of the right cerebellar hemisphere demonstrated NAA reduction, compatible with neuronal depletion, without lactate peaks. Laboratory tests and genetic investigations (including standard karyotype, CGH-Array, and FMR1, ATM, MRE11 and APTX gene analysis) showed no abnormities. Auditory brainstem responses, visual evoked potentials, somatosensory evoked potentials at upper and lower limbs, nerve conduction velocity and electromyography were normal.
A first awake EEG, performed at the age of seven years, showed physiological structured background activity with spikes and spike-and-wave complexes over posterior regions during wakefulness, in addition to generalized spike-and-wave complexes, isolated and in brief bursts during sleep, in the absence of clinical events. Seizures appeared at the age of eight years and six months with right focal motor to bilateral tonic-clonic events during sleep, treated successfully with levetiracetam.
Molecular diagnosis of de novo ADCK3 mutation (c.901C>T;c.589-3C>G) was performed at the age of 13.5, and treatment with CoQ10 (Ubidecarenone, 15 mg/kg three times per day at meal times) was started, with no further motor deterioration. Neuropsychological evaluation performed using the Wechsler Intelligence Scale for Children (4th edition) showed an IQ of 44.
At the age of 14, rare focal to bilateral tonic-clonic seizures occurred during sleep, frequently concomitant with the menstrual cycle, and the levetiracetam dose was adjusted. No seizures during wakefulness and no photosensitivity were clinically reported by the girl or her parents. A new EEG showed physiological background activity with persistent bilateral independent occipital spikes and polyspikes, activated during intermittent photic stimulation (IPS) with Grade 3 PPR, particularly during stimulation at middle-high frequency (10-30 Hz) (figure 1I). No family members had a history of photosensitivity, however, they were not tested on EEG.
Neurological examination at last follow-up visit, at 15 years, was stable. Very rare seizures during sleep were reported (once or twice a year) and EEG showed a reduction in PPR to Grade 1 (figure 1J).
Discussion
ADCK3 autosomal recessive ataxia is characterized by variable degree of cerebellar atrophy and a broad phenotypical spectrum. Since the first reports, 52 subjects harbouring ADCK3 mutations have been published: 18 (34.6%) had epilepsy, controlled by antiepileptic drugs in 10/18 cases. Focal to bilateral tonic-clonic seizures were described as the most frequent phenotype, one third of cases (6/18) experienced EPC and status epilepticus occurred in four patients, all displaying stroke-like lesions on brain MRI (table 1).
The mechanism of epileptogenesis associated with ADCK3 mutation is not known, but mouse models lacking COQ8A demonstrated increased seizure susceptibility after pentylenetetrazole injection, showing occasional seizures during daily manipulation [8]. Here, we report the EEG features of PPR in an ADCK3-mutated female adolescent. To the best of our knowledge, clinical or EEG response to IPS has never been described in epileptic patients with ADCK3 mutation nor in animal models.
Photosensitivity is defined by presence of typical encephalographic traits, known as PPR, when IPS is applied. According to Waltz's classification of 1992, PPR ranges from Grade 1 (indicating spikes within the occipital rhythm) to Grade 4 (generalized spikes and waves) [9]. The majority of subjects known to have epilepsy show PPR Grade 3 to 4. PPR is associated with clinical symptomatology in approximately 75% of subjects, depending on age, gender and presence of associated conditions, and is more frequent in adolescent females and subjects suffering from neuropsychiatric disorders [10].
Advances in neurology, basic neurophysiology and genetics have broadened our knowledge of PPR, but pathophysiological mechanisms remain poorly understood. The involvement of the occipital cortex (but also frontal and parietal cortices), as well as deep brain structures which predispose to photosensitivity, have been studied [11]. Moreover, the modulatory role during photic stimulation of the cerebellum, of which deep structures are known to be involved in drug-resistant focal and generalized epilepsies during sleep [12], has been reported both in experimental and human studies [13].
Animal models of photosensitivity in Papio papio (injected with pentylenetetrazole) have demonstrated different influences of cerebellar disruption on photosensitivity, depending on lesion location; hemispheric lesions frequently caused a transient increase in PPR response up to Grade 4, while vermian lesions induced reduction of photosensitivity with increase in non-epileptic myoclonus [14]. Moreover, studies on photosensitive baboon models showed a decrease in cerebellar activity (also in the occipital lobes and posterior cingulate gyri) during ILS on PET imaging [15]. In this view, the PPR observed in our patient could be linked to the impairment of cerebellar activity associated with photosensitivity due to cerebellar atrophy.
Interestingly, PPR has been reported in a patient with polymerase gamma (POLG) disease and epilepsy [16]. Of note, the ADCK3 epileptic phenotype shares some similarities with that of POLG-related diseases, since they are both frequently associated with SLE, occipital/posterior epilepsy, EPC, status epilepticus, migraine and migrainosus status [6]. Moreover, abnormal response to IPS mostly at low frequencies, is an early marker and prognostic indicator in patients with other progressive neurodegenerative disorders such as neuronal ceroidlipofuscinoses. This has been reported in Lafora's disease, Unverricht-Lundborg and, remarkably, mitochondrial disorders such as myoclonus epilepsy and ragged red fibres (MERRF) [17].
Considering the role of the cerebellum in modifying PPR, as well as the presence of PPR upon low-frequency IPS in other genetic progressive conditions and the trend of increased brain excitability in ADCK3 patients, we may hypothesise that photosensitivity is likely to be underestimated in these subjects, since it has not been systematically investigated in previous cases.
Substitutive therapy with CoQ10 has led to controversial results, however, early diagnosis would appear to allow for early treatment with better motor outcome [5, 6, 18]. Similarly, our patient showed a mild progressive course during the first years after onset, with no further worsening of motor involvement after the introduction of CoQ10, while sporadic seizures persisted. However, the EEG performed after 1.5 years of therapy with CoQ showed a decrease in PPR grade (figure 1). This mild EEG amelioration supports the hypothesis of Hikmat et al.[6]., of a potential role of ubiquinone in the treatment of ARCA2 disorders, not only for cerebellar symptoms, but also for epileptic activity [5, 6, 18].
Larger prospective studies are needed to understand the prevalence PPR in ADCK3-related epilepsy and its potential role as a marker of neuronal degeneration as well as therapeutic response to ubiquinone.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
The authors acknowledge Maria Teresa Dapelo and Tiziano Prastaro for technical EEG support and the patient's family for being willing to publish this report. The DINOGMI contributed to this work within the framework of the DINOGMI Department of Excellence of MIUR 2018-2022 (law 232/2016).
Two authors (EB, GZ) are members of the European Reference Network for Rare Neurological Diseases - Project ID No 739510.
None of the authors have any conflict of interest to declare.