Epileptic Disorders
MENUPhenotypic spectrum and long-term outcome of children with genetic early-infantile-onset developmental and epileptic encephalopathy Volume 24, numéro 2, April 2022

- Mots-clés : phenotypic spectrum, long-term outcome, genetic, early-infantile-onset DEE, therapy
- DOI : 10.1684/epd.2021.1394
- Page(s) : 343-52
- Année de parution : 2022
Objective. Developmental and epileptic encephalopathy (DEE) is characterized by refractory seizures, developmental delay or intellectual disability, which may be caused by gene mutation. In this study, we explored the clinical phenotype and long-term outcome in children with genetic early-infantile-onset DEEs.
Methods.Next-generation sequencing was performed on 470 patients diagnosed with early-infantile-onset DEE between 2010 and 2020. The genetic variation in all cases was classified and evaluated to identify pathogenic variants. The identified variants were further verified by Sanger sequencing.
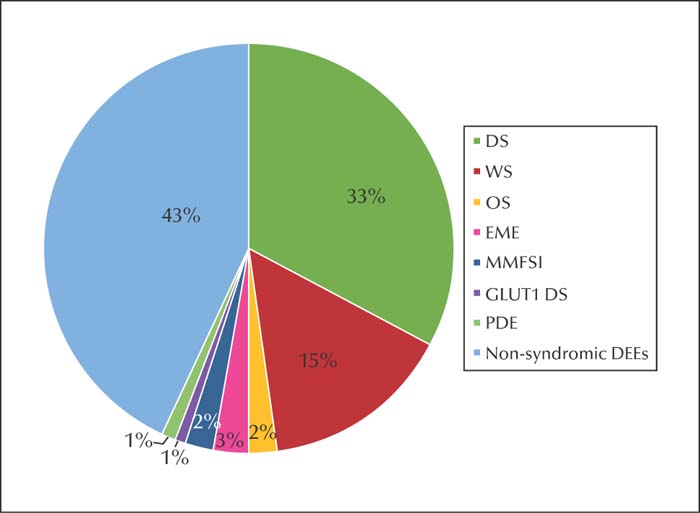
Results.A total of 118 and 10 patients were found to have putative disease-causing gene mutations and copy number variations, respectively. SCN1A mutations were detected in 38 patients (38/118, 32.2%), representing the largest proportion. In patients with early-infantile-onset DEE with burst suppression, KCNQ2 mutation was found in six patients, and the remaining mutations were reported in SCN2A (n=2) and STXBP1 (n=1). Seven patients with dyskinesia were described. In patients with non-syndromic genetic early-infantile-onset DEEs, we detected possible rare pathogenic variants in SETBP1, DPYD, CSNK2B,and H3F3A. With regards to inheritance pattern, de novo heterozygous mutations accounted for the majority (104/118; 88.1%). Three patients with SMC1A mutations responded well to ketogenic diet add-on therapy. Addition of valproic acid showed good therapeutic effects against KCNB1 and PACS2 encephalopathy.
Significance. We detected four possible rare pathogenic gene variantsas non-syndromic genetic causes of early-infantile-onset DEEs. Although early-infantile-onset DEEs responded poorly to antiseizure medication treatment, we found that specific antiseizure medications showed good therapeutic effects in some patients with early-infantile-onset DEEs harbouring gene variants.