Epileptic Disorders
MENUOutcome of status epilepticus. What do we learn from animal data? Volume 16, numéro spécial 1, October 2014

Status epilepticus (SE) was defined by the ILAE in 1993 as a condition in which a single seizure, or more than one seizure, continues for >30 minutes without recovery of function/consciousness (Dodson et al., 1993). Several definitions have been proposed and published over the last 50 years to define SE. One of the critical points in these definitions is the criterion of time, and two time periods corresponding to the duration of SE are usually considered: 20-30 minutes and 5-10 minutes. An operational definition has been more recently proposed using a cut-off of 5-10 minutes for prompt initiation of treatment (Lowenstein et al., 1999). This definition was based on the fact that a single seizure rarely lasts for longer than 2-10 minutes. After 10 minutes of duration, a seizure might last for about 30 minutes (Shinnar et al., 2001). Initially, the criterion of a 20-30-minute duration was based on the occurrence of neuronal damage and the initiation of systemic effects, potentially damaging the central nervous system. Several experimental studies have been essential in helping clinicians describe SE, and since these early initial studies (Lothman, 1990; Meldrum et al., 1973; Nevander et al., 1985), further experimental studies have helped us to better understand the consequences of SE. Our aim in this review is to firstly define what kind of animal models are helpful to further understand the consequences of SE and secondly, to discuss data from experimental studies that are helpful in our understanding of this condition.
What is a good animal model?
Much of our knowledge of epilepsy is based on the use of animal models. Animal models in the field of epilepsy are useful for a variety of tasks: the investigation of pathophysiological mechanisms, the evaluation and development of new antiepileptic treatment, and the study of the consequences of conditions that may be concurrent with epilepsy (cognitive consequences and/or comorbidities).
Animal models should meet some criteria that have been previously suggested: (1) the animal model should exhibit similar electrophysiological correlates/patterns observed in the human condition; (2) aetiology should be similar (genetic predisposition, injury, etc.); (3) the proposed animal model should be scaled or reflect a similar age in humans at which time the manifestations of the epilepsy syndrome are age-specific; (4) the animal model should display similar pathologies to a human condition which exhibits specific pathological changes (e.g. cortical dysplasia); (5) the condition being modelled should respond to similar antiepileptic drugs; and (6) the behavioural characteristics (short- or long-term behavioural changes) should reflect behavioural manifestations observed in humans (Auvin et al., 2012).Criteria to evaluate an animal model
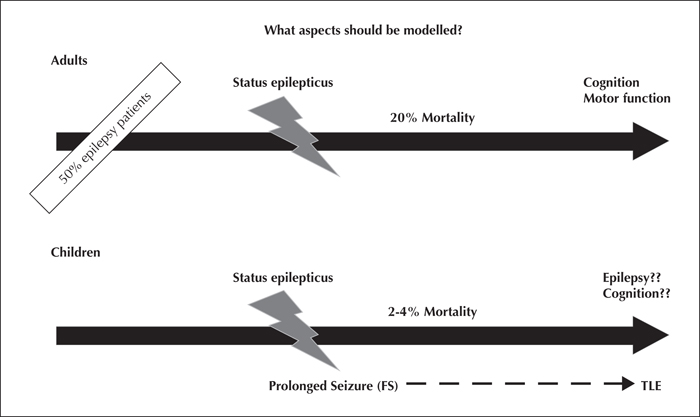
What aspects should be modelled?
The overall incidence of SE is about 40/100,000 persons per year (DeLorenzo et al., 1996). In humans, there are several differences between SE that occurs in adults and children (figure 1). In adult patients, the mortality is high (10-25%) (Logroscino et al., 2005) but the incidence is lower than in childhood (DeLorenzo et al., 1996; Hesdorffer et al., 1998; Chin et al., 2006). The mortality in children is about 2-4% (DeLorenzo et al., 1996). Regarding clinical data, it would appear that outcome is not clearly related to either duration or frequency of SE (Metsaranta et al., 2004), but predominantly related to the underlying aetiology (Sillanpaa and Shinnar, 2002; Stroink, 2007; Camfield and Camfield, 2012).
The number of studies in which an attempt was made to address the risk of epilepsy after SE is limited. Moreover, the results are highly variable with an overall risk of between 15 and 27% in prospective studies, while the estimated risk in retrospective studies is reported to reach 50% (Annegers et al., 1982; Maytal et al., 1989). It appears that most of the children who develop epilepsy following SE have a pre-existing neurological disorder or neurological risk factor (Maytal et al., 1989; Shinnar et al., 1996). A role for an underlying lesion in the pathogenesis of sequelae of SE, including the occurrence of epilepsy, has been realistically suggested.
Status epilepticus in the mature brain
In rodents, SE can be induced by injection of chemicals or by electrical stimulation. There are now several models that are well described: injection of kainic acid (Benari, 1985), injection of pilocarpine (with or without lithium) (Turski et al., 1986), continuous electrical stimulation of the hippocampus (Bertram, 1997), and continuous electrical stimulation of the amygdala (Nissinen et al., 2000).
In these various models, seizures initially originate from limbic structures followed by secondary generalisation. The seizures are long-lasting, progressing to SE with relatively high mortality (Stafstrom et al., 1993; Nissinen et al., 2000). There is a rapid alteration in neuronal networks occurring within minutes to days following SE. These changes are related to modifications of ion channel kinetics, related to membrane depolarisation and modification of protein function by post-transriptional regulation and early gene activation. The initial changes are followed by subacute changes over weeks. This step includes transcriptional events, neuronal death, and inflammation. All these processes result in anatomical changes inducing network reorganisation (mossy fibre sprouting, gliosis, neurogenesis, etc.). All these phases lead to the development of spontaneous recurrent seizures (SRS) (Stafstrom et al., 1993; Priel et al., 1996; Nissinen et al., 2000). These different steps of epileptogenesis are extensively studied in order to understand the role of the various factors leading to SRS. This may therefore provide a basis with which to develop a new therapeutic approach to prevent epileptogenesis and discover biomarkers in order to identify patients who are at risk of developing epilepsy (Rakhade and Jensen, 2009).
SE induces neuronal cell loss. The SE-induced cell injury is widespread. In the hippocampus, this is observed in CA-1, CA-3, and dentate regions. Cell injury has been also reported in the amygdala, piriform, and entorhinal cortex and thalamus (Bertram, 1997; Nissinen et al., 2000). Based on experimental models of SE in adults, some studies show that the severity of the cell loss correlates with seizure duration (Benari, 1985; Covolan and Mello, 2000). In humans, there are very few reports (<20) of human pathological studies after SE (Tsuchida et al., 2007). Based on imaging studies, patients generally present evidence of oedema, at least in the temporal cortex, with neuronal necrosis observed later in the same areas. These few human studies appear to support the findings of animal models of SE, showing direct excitotoxic injury in the absence of hypoxia-ischaemia.
After a latent period following the SE attack, most rats have SRS, including secondary generalised clonic seizures (Stafstrom et al., 1993; Priel et al., 1996; Nissinen et al., 2000). The loss of neurons in the hippocampus has been suggested to play a role in SRS, in particular, the loss of inhibitory interneurons (Cossart et al., 2001; Brandt et al., 2003). In addition, the animals show significant learning and memory deficits (Nissinen et al., 2000; Detour et al., 2005). These deficits appear to be likely related to SE-induced brain damage in limbic areas.
Status epilepticus in the immature brain
Animal studies using rodents have suggested that the developing brain is less vulnerable to SE, compared to the mature brain. There is far less cell loss and epileptogenesis following SE in models of immature rats, relative to adult rats.
Rats are commonly used in epilepsy research of the developing brain. The multitude of differences in the rate of maturation between human and animal brains are well recognised, making precise comparisons at equivalent developmental milestones between the two species a multidimensional task. Using various parameters, we use the following as a reference for maturation- seventh postnatal day (P7) to P10: term human newborn; P14-P21: toddlers to young children; P28: older children prior to puberty; P35: adolescent.
Status epilepticus
As reported in adult rats, the administration of kainic acid (KA) or the administration of pilocarpine or lithium-pilocarpine represents the most widely used epilepsy models of SE in immature rats (Benari, 1985; Turski et al., 1986).
Using KA, immature rats at P15 cannot survive at doses tolerated in adults. Systemic KA (3 mg/kg) in P15 rats did not produce a significant level of cell injury, even though severe seizures occurred (Albala et al., 1984; Okada et al., 1984). The extent of cell injury is highly variable according to the route of administration of the KA. An intracerebroventricular application of KA results in more severe damage than that given intraperitoneally (Montgomery et al., 1999; Humphrey et al., 2002). The mortality rate in the KA model of SE is a real problem. This rate is about 90% in P15 rats (Koh et al., 1999). Even though this mortality rate poses a problem with regards to performing an experimental study, it has been shown that 3 mg/kg of KA results in severe seizure, but no hilar or CA3 damage is observed (Okada et al., 1984). Although SE induced by KA in P15 rats does not induce identifiable brain injury, the rats that are subject to a new challenge by KA at P45 experience more severe brain damage and performed worse in spatial learning tasks, relative to controls (Franck and Schwartzkroin, 1984). Thus, although cellular damage may not be clearly demonstrated after kainic acid treatment at P15, one may not conclude that this treatment has had no effect on the immature brain.
In the lithium-pilocarpine model of SE, the level of cell injury is minimal before P14 (Sankar et al., 1998). A significant level of cell injury is observed after P14. SE-induced cell injury is maximal in CA-1 at P14, whereas only a few damaged neurons are detected in the CA3 region. At P21, both CA-1 and CA-3 regions show extensive damage (Sankar et al., 1998). Neuronal damage in the amygdala and dentate granule cells is also age-specific, and the vulnerability progressively increases with age (Sankar et al., 1998).
The occurrence of SRS after SE is variable according to the model of SE and the age at occurrence of SE (table 1). Whether cell injury to the immature brain is required to induce epileptogenesis remains an active topic of debate (Dudek et al., 2010; Baram et al., 2011). In the lithium-pilocarpine model, the occurrence of SRS in adulthood is observed in 10-20% of rats that underwent SE at P14 (mostly stage 1-2 of the Racine Seizure scale) and 50% of rats that underwent SE at P21 (Sankar et al., 1998). More recently, a study suggested that the consequences of SE in the immature brain occurred progressively and a similar level of cognitive consequences and epileptogenesis, compared to more mature brain, was reached (Kubova and Mares,2013). Using the lithium-pilocarpine model at P12, they reported that both the severity and incidence of SRS tended to progress with time (50% at 5 months after SE and 87.5% at 7 months after SE). Rats that experienced SE at P25 were monitored at 5 months after SE and seizures were detected in 83.3% of animals. Cell count of hippocampal neurons performed after video-EEG monitoring revealed loss of hilar neurons in both age groups. In P12 rats, morphological damage also tended to progress over time (Kubova and Mares,2013). This recent study suggests that the animals that become epileptic after having undergone SE, at a time when the brain is immature, have been previously underestimated.
Double-hit injury models
In addition to the model of SE, different types of injury and/or prolonged seizures have been used to induce epileptogenesis, with the goal of mimicking human conditions (table 1). Hyperthermic seizures induced in postnatal-day-10 or -11 (P10-P11) rat pups are used to mimic prolonged febrile seizures (Toth et al., 1998). Hypoxic-ischaemic brain insults can also be induced in rat pups to model hypoxic-ischaemic encephalopathy, which is a prominent cause of mortality in neonates and morbidities, including epilepsy, in children. Animal models involving hypoxia in P7 to P10 rats have been used to study both acute neonatal seizures and the subsequent development of epilepsy (Kadam et al., 2010; Rakhade et al., 2011).
The use of models with a double hit provides data that allows us to further understand the contribution of early-life events in the future development of epilepsy. The motivation for developing “two-hit” animal models was to include a clinical relevant component to prolonged early-life seizures. As an example, a prolonged febrile seizure often precedes the development of temporal lobe epilepsy with mesial temporal sclerosis (Cendes, 2004). In order to evaluate the role of inflammation as a component of febrile seizures in children (Auvin and Vallee, 2009), we recently conducted a study using a double-hit injury approach (SE+systemic inflammation). We have reported that inflammation increases SE-induced cell injury at both P7 and P14 (Auvin et al., 2007). The combination of systemic inflammation to P14 SE results in an increase of epileptogenesis (Auvin et al., 2010). Similarly, the assessment of the role of focal cortical dysplasia in the occurrence of prolonged seizures in the developing brain is possible in some experimental studies. Using rat pups that had a right sensorimotor cortex freeze lesion at P1, followed by hyperthermic seizure at P10, the rats with a localised freeze lesion had a lower seizure threshold and experienced prolonged ictal manifestations suggesting that focal cortical lesion is involved in atypical febrile seizures (Scantlebury et al., 2004). Moreover, this pre-existing cortical lesion modified epileptogenesis following a prolonged seizure. The same experimental group reported that 86% of patients with the lesion plus the hyperthermic seizure group experienced development of SRS, while none of the controls exhibited any abnormality (Scantlebury et al., 2005).
Effect of anticonvulsant treatment and anti-epileptogenesis treatment
The use of antiepileptic drugs to stop SE in humans has been evaluated in the model of SE. The cessation of SE by diazepam decreases the level of SE-induced cell injury (Pitkanen et al., 2005; Francois et al., 2006). The use of 2.5 mg/kg of diazepam in adult rats stops seizures and decreases the mortality rate. In addition, the cessation of seizures by diazepam results in a lower level of cell loss in CA-1, in the hilus, and in the piriform cortex, compared to controls (Francois et al., 2006). When SE is stopped by diazepam (20 mg/kg) 2 hours or 3 hours after SE, the outcome is modified. The rate of animals that develop epilepsy after SE was reduced when diazepam was given to stop SE. The percentage of epileptic animals was lower (42%) in the 2-hour DZP group, compared to the 3-hour DZP group (71%). Moreover, the seizures were less frequent in the DZP-treated group, with a marked reduction when treatment was given after 2 hours of SE (Pitkanen et al., 2005).
Despite vast laboratory evidence that many anticonvulsant medications possess neuroprotective properties (Pitkanen, 2002; White, 2002), pharmacological strategies to mitigate or prevent epileptogenesis in humans have failed (mostly in head trauma) (Holtkamp and Meierkord,2007; Loscher and Brandt,2010). This failure might be related to several issues such as species differences, inappropriate timing of intervention, focus on the wrong molecular targets, differential genetic susceptibility, and perhaps long-term neurotoxicity of drugs (Pitkanen and Lukasiuk, 2011). It has been suggested that the molecular targets that have traditionally been studied may be more relevant to ictogenesis (i.e. induction of an acute seizure) rather than epileptogenesis (Kobow et al., 2012).
Conclusion
Animal models of SE have provided a better understanding of the diversity of behavioural, structural, and functional changes resulting from SE. It is now well established from the experimental studies that the consequences of SE are mainly related, but not entirely, to the developmental age when SE occurs. Regarding the criteria of animal models (see above), a good model of the acute stage of SE is provided. However, it should be stated that none of the models using initial injury to induce epileptogenesis can be considered as models of paediatric epilepsy, because the occurrence of SRS during adulthood is always reported (Auvin et al., 2012).
It is obvious that animal studies have also inherent limitations; SE is induced by the administration of noxious agents and is not spontaneous. The experimental data show that the outcome appears to be species- or model-specific. It is currently difficult to draw any definitive statements regarding model specificity because the data described in the literature are often confounded by differences in the route of administration of the drug, strain of rat used, and duration of SE.
Despite these limitations, animal models of SE support the notion that SE induces brain damage. The younger the age at the time of SE, the less severe the consequences appear to be. However, several factors, including the cause of SE (evaluated by double-hit injury models), modify the seizure severity as well as the level of injury and related epileptogenesis.
The use of treatment to stop the seizure and/or the duration of the SE results in a decrease of SE-induced cell injury. Preliminary data suggest that anti-epileptogenesis treatment with a single target is limited, while multiple targets should be further explored in order to reach the realistic goal of an anti-epileptogenesis treatment (Kobow et al., 2012).
Acknowledgements and disclosures
Stéphane Auvin is partially supported by INSERM Grant (Contrat Interface INSERM 2010).
None of the authors has any conflict of interests to disclose.