Epileptic Disorders
MENUMild malformation of cortical development with oligodendroglial hyperplasia and epilepsy (MOGHE): a widespread disease with an apparently focal epilepsy Volume 23, numéro 2, April 2021

Mild malformation of cortical development with oligodendroglial hyperplasia and epilepsy (MOGHE) is a recently recognized, highly epileptogenic, distinct histopathological entity in refractory epilepsy that primarily involves the frontal lobes [1]. Very few cases have been reported in the literature. Surgical outcomes are variable. MOGHE is based on a histopathological diagnosis, but recognition during presurgical evaluation aids prognostication and surgical planning. We discuss the clinical, electrographic, radiological and histopathological features of MOGHE through two illustrative cases.
Case 1
A 23-year-old, right-handed male patient with drug-resistant epilepsy (DRE) since 14 years of age underwent pre-surgical evaluation. His birth and development were normal but he performed below average scholastically. He had daily, mostly nocturnal, seizures lasting 1-2 minutes. He had status epilepticus twice, requiring ventilation and anaesthetic agents.
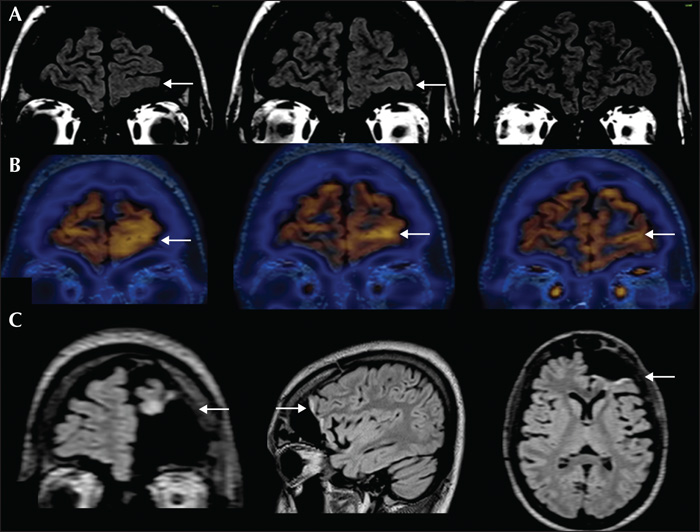
During video-EEG, the recorded seizures were characterized by behavioural arrest, restlessness, and tonic facial contraction, followed by asymmetric (right more than left) tonic posturing of the limbs and trunk, pedalling movements and hypermotor behaviour. Postictally, he had restlessness, a wandering tendency and was amnestic to the event. He reported an aura of vague cephalic sensation in some seizures. Frequent interictal epileptiform discharges (IEDs) were seen over the left frontal, left frontopolar, left frontotemporal and bifrontal regions (supplementary figure 1A, B). Ictal EEG showed generalized suppression without any clear lateralization (supplementary figure 1C). Brain MRI (figure 1A) showed left fronto-polar and orbitofrontal loss of grey-white differentiation, suggesting focal cortical dysplasia (FCD). FDG-PET showed a corresponding area of hypermetabolism (figure 1B). Neuropsychological assessment showed diffuse cognitive dysfunction.
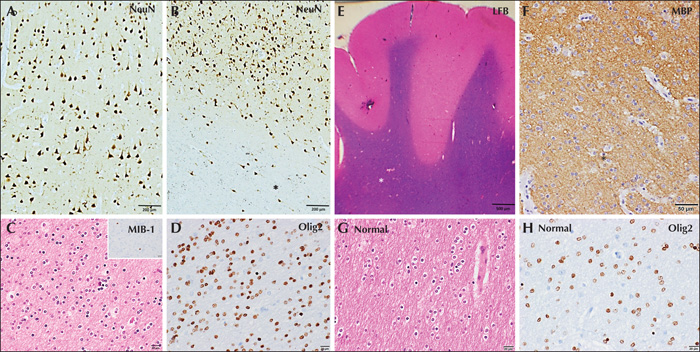
He underwent electrocorticography (ECoG)-guided surgery. ECoG showed frequent, often rhythmic, spikes and polyspikes as well as intermittent delta brush and brief ictal rhythms from the orbitofrontal and ventrolateral prefrontal regions (supplementary figure 2A, B). Post-resection, ventrolateral frontal areas showed persisting IEDs without electrographic seizures. Further resection was deferred due to proximity to the language area. Histopathology showed normal cortical laminar architecture with subcortical heterotopic neurons highlighted by NeuN. Dysmorphic neurons and balloon cells were negative for phosphorylated neurofilament, GFAP and vimentin. The white matter showed a striking increased density of oligodendroglial cells labelled with Olig2, with prominent perivascular clustering. Luxol fast blue and myelin basic protein stains showed hypomyelination in areas with increased oligodendroglial cell density. A few of the oligodendroglial cells in the white matter were labelled by the proliferative marker, MIB-1. All markers for diffuse glioma (IDH1 [R132H], ATRX, and p53) were negative. Hence, the histopathological features were considered to be consistent with the diagnosis of MOGHE (figure 2). AED tapering, three months post-surgery, resulted in a cluster of seizures. MRI (figure 1C) showed adequate resection with regard to the initial MRI abnormality. Subsequently, the patient had no further seizures and at the last follow-up visit, at two years, he was leading an independent life.
Case 2
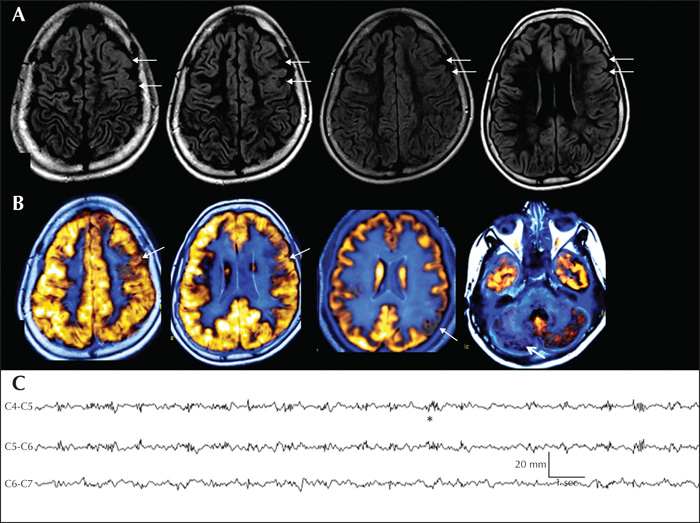
A 14-year-old male child with a history of mild global developmental delay and seizure onset at nine months of age was evaluated for refractory epilepsy. He had 8-10 seizures daily. Video-EEG recorded tonic seizures consistent with left frontal ictal onset. IEDs were multifocal, but predominantly over the left frontal region. Also noted was left frontal-temporal theta slowing. Brain MRI (figure 3A) showed increased cortical signals with loss of grey-white differentiation in the left frontal region but the extent was unclear. Interictal FDG-PET showed widespread hypometabolism involving the left frontal and parietal regions, maximally corresponding to the MRI abnormality (figure 3B). PET hypometabolism was also noted in the right cerebellum, suggesting crossed hypometabolism (figure 3B). Neuropsychology showed diffuse dysfunction, maximally involving frontal functions. Functional-MRI lateralized language to the right hemisphere. Intraoperative ECoG showed frequent spikes, polyspikes and delta brush overlying the area of radiological abnormality (figure 3C). Electrographic seizures were recorded from left prefrontal areas. The patient underwent left frontal resection. Histopathology revealed striking oligodendroglial hyperplasia in the white matter labelled by Olig2. The overlying cortex showed preserved cortical lamination but with subcortical heterotopic neurons consistent with MOGHE. The white matter showed hypomyelination in areas with increased oligocytosis, as highlighted by Luxol fast blue and myelin basic protein immunohistochemistry. Scattered oligodendrocytes were labelled by MIB-1, reflecting a proliferative cycle. Five years post-surgery, the patient was classified with Engel Class III outcome.
Discussion
MOGHE was first described in a case series of 22 patients [1]. All had frontal lobe epilepsy with MRI features of FCD. Histopathology showed increased Olig2 and PDGFR alpha-immunoreactive oligodendroglia in white matter and deep cortical layers with heterotopic neurons in the white matter.
We analysed the electroclinical, radiological and pathological features of our cases with MOGHE. Both had childhood-onset, highly refractory, frontal lobe epilepsy with daily seizures, one of whom had recurrent status epilepticus. Both had significant learning difficulties and diffuse cognitive dysfunction.
Hartlieb et al. described age-related MRI variations in MOGHE [2]: Subtype I in early life, with increased laminar T2 and FLAIR signals at the corticomedullary junction (CMJ) and Subtype II in adolescents, characterized by reduced differentiation of CMJ because of increased adjacent white matter signals. Both our patients had Subtype II of MOGHE. Brain MRI with an epilepsy protocol was performed in both patients (3T-MRI in Case 1 and 1.5T in Case 2) to include T1, T2, FLAIR, gradient, and DTI imaging with added 3D-T1 and 3D-T2 sequences. 3D-FLAIR sequence was available in Case 1. High-resolution MRI is recommended for identification of features suggestive of MOGHE which can be missed with non-availability of 3D sequences. Difficulty in defining the margins in MOGHE affects surgical planning, but higher MRI resolution may provide better delineation of the lesion [3].
There are limited reports on electrographic characteristics of MOGHE. Garganis et al. described two cases of temporal plus epilepsy with widespread IEDs [4]. Our patients also had frequent multifocal and widespread IEDs, disproportionate to the MRI findings. Ictal EEG onset was diffuse and non-localizing, similar to other frontal lobe epilepsies. Therefore, presurgical localization was weighted on semiology and imaging findings rather than electrography.
FDG-PET abnormalities in MOGHE can mimic FCDs. In Case 1, interictal FDG-PET showed left frontal hypermetabolism which is consistent with FCD [5]. The hypermetabolism is likely a reflection of persistent ictal activity. In Case 2, more widespread hemispheric and crossed cerebellar hypometabolism were seen.
ECoG-based resections are usually adequate in frontal lobe epilepsies when MRI shows a concordant FCD. ECoG of FCDs is characterized by repetitive electrographic seizures, repetitive bursts of discharge, continuous or quasi-continuous rhythmic spiking and a delta brush pattern [6, 7]. Both our patients had ECoG patterns consistent with FCD. Post-resection ECoG showed persistent IEDs, again suggesting a widespread pathology.
Surgical outcomes in MOGHE have been variable.Schurr et al. reported seizure recurrence in 61% of 22 operated patients while 33% were free of disabling seizures [1]. Recently, more favourable surgical outcome has been observed. Hartlieb et al. in 2019 showed (Engel Class 1 and II) outcomes in 16/25 of their patients [2]. Garganis et al. described two temporal plus MOGHE patients, one was rendered seizure-free while the other had a poor outcome [4].Verentzioti et al. reported a patient who was seizure-free at two years after surgery [8]. One of our cases had Engel Class 1D outcome but the other had an Engel Class III outcome. Variable surgical outcomes could be due to the widespread nature of MOGHE and surgical restrictions by eloquent cortices.
Histopathology remains the gold standard for the diagnosis of MOGHE and has been described in detail [1]. Histopathology in our patients showed normal cortical ribbon on NeuN immunohistochemistry (IHC) with an increase in density of heterotopic neurons in subjacent white matter. Dysmorphic neurons and balloon cells were negative for phosphorylated neurofilament, GFAP and vimentin. An increase in the subcortical white matter oligodendroglial population was noted, with few cells labelling for MIB-1. Luxol fast blue and myelin basic protein stains for myelin revealed hypomyelination in areas with increased oligodendroglial cell density. The differential diagnosis of MOGHE includes oligodendroglial hamartoma and oligodendroglioma. Hamartomas are characterized by a lack of proliferative activity. In both our cases, immunohistochemical markers for oligodendroglioma were negative, though FISH for a 1p/19q co-deletion study was not performed. A higher level of oligodendroglial proliferation was noted in younger patients with MOGHE [1].Neuronal activity has been shown to promote oligodendrogenesis [9].
Different patterns of oligodendroglial proliferation have been observed in other instances of DRE [10, 11]. The pathogenesis of oligodendroglial hyperplasia in MOGHE is unclear. Further studies are required to understand the role of oligodendroglial hyperplasia in DRE and MOGHE, and in particular, to understand the natural history of the disease.
Conclusion
MOGHE is a recently described disorder which usually presents as highly refractory childhood-onset epilepsy with predominant frontal localization. The seizures appear focal, however, widespread electrographic and PET abnormalities disproportionate to the MRI, cognitive impairment and lower rates of seizure freedom after surgery suggest a more widespread pathology. Some of these pre-surgical characteristics of MOGHE could help presurgical planning for better outcomes.
Supplementary data
Supplementary figures are available on the www.epilepticdisorders.com website.
Disclosures
None of the authors have any conflict of interest to declare.