Epileptic Disorders
MENUMajor intra-familial variability in Unverricht-Lundborg disease Volume 24, numéro 1, February 2022

- Mots-clés : Unverricht-Lundborg, psychiatric, phobia, late-onset, familial variability
- DOI : 10.1684/epd.2021.1372
- Page(s) : 163-70
- Année de parution : 2022
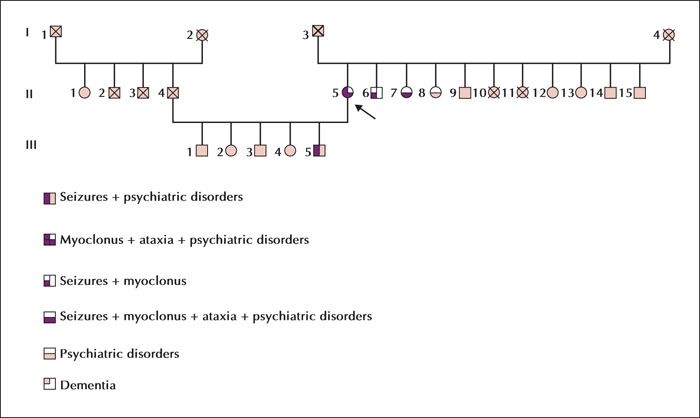
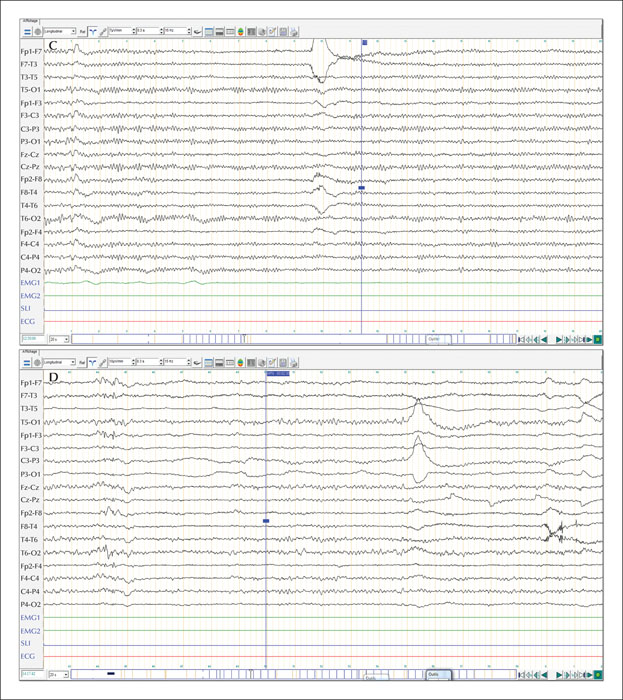
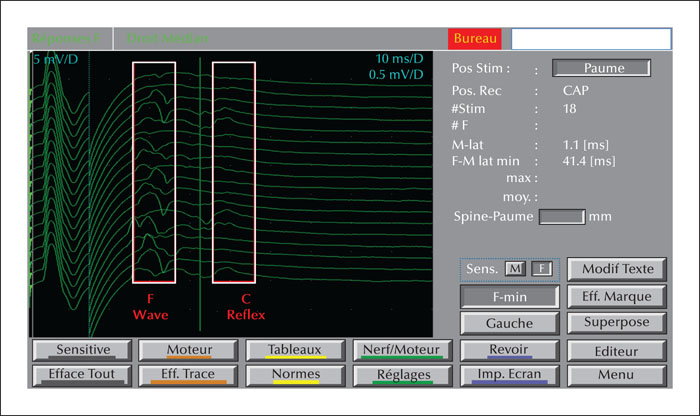
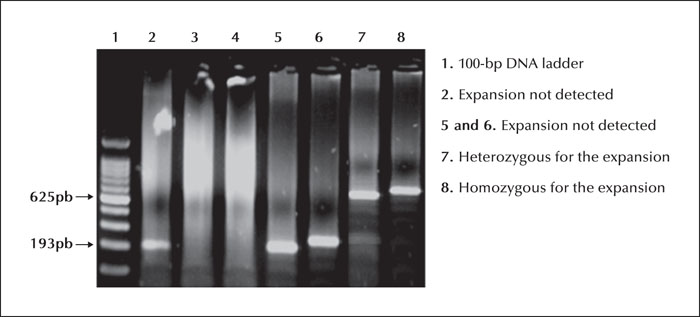
Unverricht-Lundborg disease (ULD), also called progressive myoclonic epilepsy type 1, is usually characterized by the presence of ataxia associated with myoclonus and epileptic seizures without progressive cognitive deficit, presenting during late childhood and early adolescence. Currently, there is a growing body of evidence for atypical presentations of the disease with a milder phenotype or without the full symptomatology. We describe a case report of a late-onset phenotype with progressive myoclonus-ataxia syndrome accompanied by initial recurrent falls, resulting in specific phobia and agoraphobia starting at the age of 50 years old. The examination revealed multifocal myoclonus with cerebellar ataxia and electroencephalogram showed generalized polyspikes and spike-wave discharges. Electromyogram revealed positive myoclonus of 60-ms duration in the face and the presence of C reflex. A genetic study confirmed the diagnosis of ULD in the patient and other additional family members, presenting a wide range of intra-familial variability. We discuss the challenging differential diagnosis for such a misleading presentation and its possible underlying pathophysiological mechanisms. Our case report may contribute to broadening the age and clinical boundaries for this disease and emphasizes the intra-familial age and symptom variability. Based on a suggestive family history, the diagnosis of ULD should be considered in this context, even in older patients.