Epileptic Disorders
MENUIntracranial investigation of a patient with nodular heterotopia and hippocampal sclerosis: dealing with a dual pathology Volume 19, numéro 2, June 2017

Clinical history
A 29-year-old right-handed female presented for management of drug-resistant epilepsy. Gestational, birth, and developmental history were normal. There was no history of significant head injury or neuroinfections, and no family history of seizures. She suffered one simple febrile seizure at the age of 3 years. Neurological examination was normal.
Seizures began at the age of 23 years and were characterized by partial loss of consciousness; she was able to respond to some questions, but was slow and sometimes incoherent. She stared into space and performed repetitive subtle movements, such as scratching her eyebrow with her right-hand fingers and mild chewing, followed by blinking and searching ocular movements, which typically lasted between one to three minutes. After the episode, she was able to recall that she had had visual illusions; she described them as “everything seems zoomed out in appearance”, “I see everything different, as if the edges of things have changed”.
Over the last year, she had one seizure per month. Seizures with secondary generalization occurred infrequently, characterized by complete loss of consciousness, deviation of the head/eyes to the left, and generalized clonic jerks. During the presurgical evaluation, she was on lamotrigine at 225 mg BID, levetiracetam at 1,500 mg BID, and clobazam at 10 mg BID without response.
Pre-surgical work-up
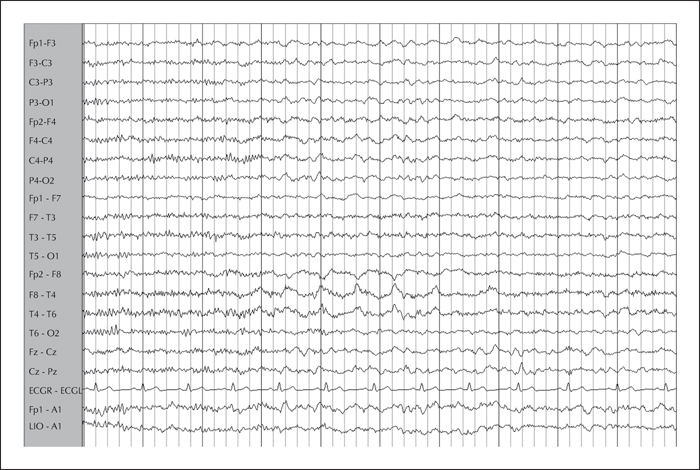
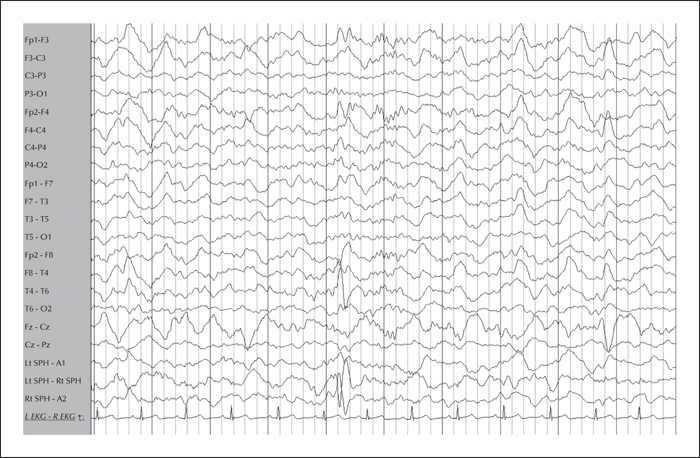
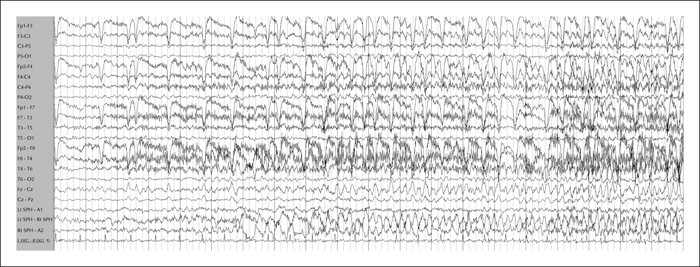
Previous EEG studies revealed normal background and symmetric sleep. A right temporal intermittent rhythmic delta activity (maximal at F8-T4) was present (figure 1). Video-EEG telemetry showed frequent spikes and sharp waves over the right temporal region (maximal at F8-T4) (figure 2). Electrographic ictal onset was localized consistently to the right mesial temporal region, with later involvement of the contralateral hemisphere during longer events (figure 3).
Her seizures were characterized by staring, behavioural arrest, and partial loss of consciousness, followed by subtle right-hand automatisms and lip smacking, sometimes accompanied by a dystonic left hand. She never reported subjective symptoms during the onset of seizures. Automatisms were followed by eyelid fluttering lasting for 1-2 minutes. Few episodes were followed by any report of late visual hallucinations (six minutes after onset), consisting of flashing lights over the left visual area and head/eye deviation to the left, left upper body turning, and rhythmic upper body jerking with secondary generalization.
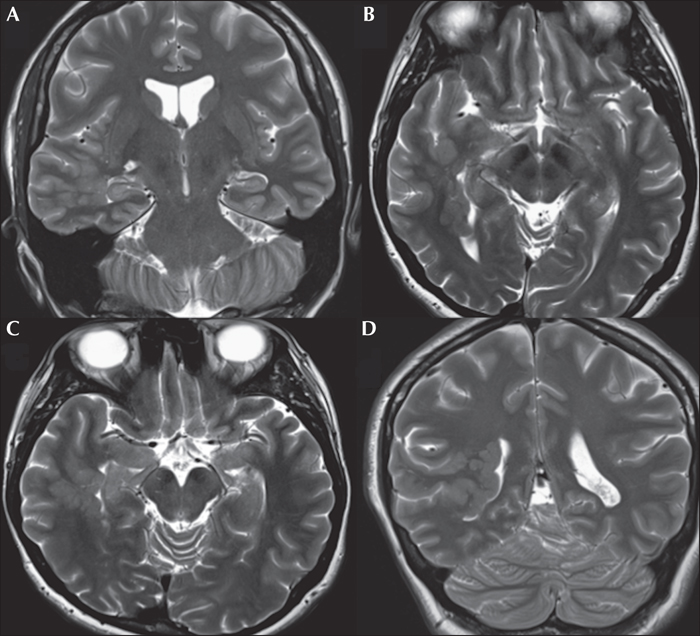
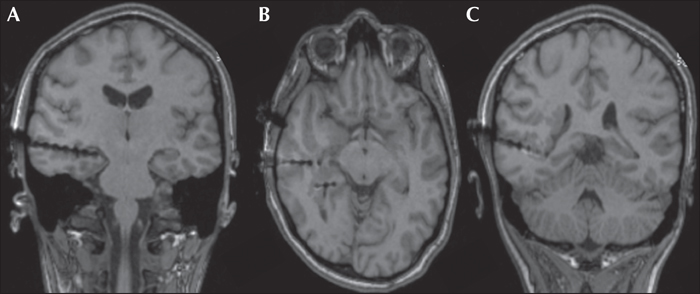
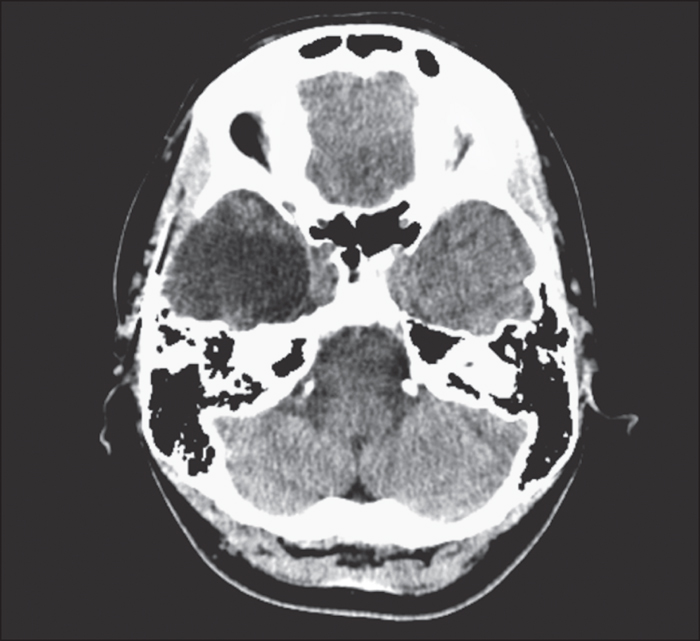
Brain 1.5T MRI showed unilateral periventricular heterotopic grey matter predominantly in the right posterior temporal region, which extended to the occipital, parietal, and insular area (figure 4). Neuropsychological assessment at age 29 demonstrated normal, full-scale verbal and non-verbal intelligence, but a specific dysfunction in visual and spatial functions suggesting non-dominant posterior quadrant dysfunction.
Hypothesis and intracranial recording
The staring, partial loss of consciousness, and right-hand automatisms were correlated to an ictal EEG onset and irritative zone in the right temporal region; the eye fluttering, left visual hallucinations, and head deviation further suggested right posterior temporal, parietal, and occipital involvement. The functional deficit zone was considered right temporal according to the EEG slowing, and posterior quadrant according to the neuropsychological evaluation. The epileptogenic zone was hypothesized to involve mesial and lateral temporal areas. However, right parietal and occipital area involvement was a concern, given the large lesion on MRI.
Depth electrode investigation was carried out during long-term video-EEG monitoring. Recordings were performed using three intracerebral depth electrodes placed orthogonally according to Talairach's stereotactic method. Antiepileptic drugs were reduced and stopped in a phased manner over five days. Three intracerebral multiple contact electrodes were denoted by the letters HH (hippocampal head), HB (hippocampal body), and PT (posterior temporal). The first two recorded mesial structures (the anterior hippocampus and the posterior hippocampus), as well as some lateral temporal nodules, and the third recorded the posterior temporal region including structures around the heterotopic nodules (figure 5).
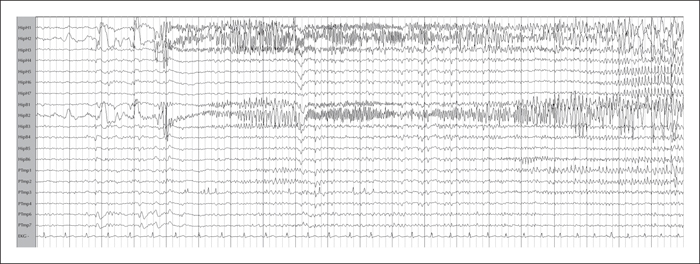
Depth electrodes revealed frequent spikes and runs of polyspikes at HH1-HB1 contacts and occasional spikes observed at PT6-7 contacts. Several 10-15-second subclinical seizures were recorded during sleep from HH1 and HB1. Five clinical seizures were registered. EEG changes began 40 seconds before the clinical onset, always characterized by sharp waves from the hippocampus (figure 6).
Surgery and outcome
During the scalp and intracranial EEG, visual symptoms were reported late, often five minutes after the clinical seizure onset. The habitual seizures started from the hippocampus, and for all the seizures, the heterotopic nodules were silent throughout the ictal period. The patient underwent a standard right temporal lobectomy, including the hippocampus and some of the heterotopic nodules (figure 7). She has been seizure-free (Engel class Ia) for three years following surgery. She is taking lamotrigine at 100 mg BID. Her quality of life has improved. Neuropathology of the tissue revealed hippocampal sclerosis (HS) and periventricular nodular heterotopia (PNH).
Discussion
We present a case of extensive PNH and HS in a patient who has been seizure-free for three years after a standard temporal lobectomy. We considered this case to be particularly interesting due to the conflicting results in the literature in terms of surgical outcomes and the number of required electrodes to investigate such patients. Heterotopic nodules exhibit variable and complex epileptogenicity and there are several reports showing seizure recurrence after limited resections (Dubeau et al., 1995; Li et al., 1997). However, less aggressive epilepsy surgery can be favourable after a careful evaluation of the seizure-generating areas (Aghakhani et al., 2005; Tassi et al., 2005; Acar et al., 2012; Agari et al., 2012).
In our case, semiology and interictal and ictal epileptic discharges suggested mesial temporal onset, but the presence of blinking, visual hallucinations, and a large lesion on MRI involving the parietal, insular, and occipital areas led to further investigations. We performed invasive monitoring with limited use of electrodes that confirmed the non-invasive work-up and showed that the hippocampus was the epileptogenic network pacemaker. Depth electrodes allowed us to limit the surgery, to remove the epileptogenic zone and leave a large part of the malformation in place.
PNH is a malformation of corticale development involving an intricate network between the nodules and the surrounding cortex. Ictal activity can start from the nodules alone, exclusively from the overlying or distant cortex, simultaneously from heterotopic nodules and neocortex, or synchronically from nodules and the ipsilateral hippocampus (Dubeau et al., 1995; Aghakhani et al., 2005; Tassi et al., 2005; Stefan et al., 2007).
The association between HS and PNH is common. Different series have reported a high percentage of dual pathology cases, from 60% (Dubeau et al., 1995; Li et al., 1997) to 100% (Raymond et al., 1995). The series of Tassi reported that only 30% of patients with nodular heterotopia have abnormalities suggesting hippocampal dysgenesis, but none had radiological or pathological signs of HS (Tassi et al., 2005). Our case did not show MRI findings suggesting HS, but the pathology confirmed the clinical suspicion.
Several authors have stated that temporal lobectomy yields poor outcome in patients with nodular heterotopia, even in those with coexistent HS (Aghakhani et al., 2005; Dubeau et al., 1995; Li et al., 1997). Li et al. (1997) reported Engel class III-IV in 100% of patients who had exclusively temporal resection with residual heterotopia. Dubeau et al. (1995) reported seizure recurrence in 70% of patients. The best predictor of seizure freedom is the detection of a focal epileptic generator demonstrated by intracranial recordings. This generator may or may not include the nodular heterotopia (Aghakhani et al., 2005; Tassi et al., 2005). In our case, as the epileptic discharges from onset involved the hippocampus and also spread to anterior nodules and neocortex (15-20 seconds), a classic temporal resection was proposed. Conveniently, the hippocampus, as well as part of the heterotopic tissue located in the anterior region of the temporal lobe, was resected during the temporal lobectomy. In this case, we used a limited number of electrodes and the most posterior electrode was useful in showing that the nodules in the posterior temporal region were not involved in the generation of the seizures, limiting the resection.
Whether a patient has left or right temporal lobe epilepsy is an essential consideration when evaluating patients for lobectomy. Left temporal resection carries an inherent risk of post-operative verbal memory loss. Fifty (Battaglia et al., 2006) to 70% (Tassi et al., 2005) of PNH patients have unilateral malformations. In 78% of these cases, the PNH is right-sided (21/27) and in bilateral asymmetric cases, the heterotopia is often more evident in the right hemisphere (78%) (Battaglia et al., 2006). The findings of the present case of unilateral right-sided PNH are consistent with those described for the majority of published cases. The non-dominant surgical nature of the case made the diagnosis slightly more straightforward.
Conclusion
This case demonstrates that the ictal onset was localized in the hippocampus despite large areas of nodular heterotopia in the same hemisphere. One should remember that every patient has a unique EEG, clinical, and radiological picture. Invasive monitoring with a reduced number of electrodes can provide adequate analysis and this is essential in planning surgical strategies.
Disclosures
None of the authors have any conflict of interest to disclose.