Epileptic Disorders
MENUGOSR2: a progressive myoclonus epilepsy gene Volume 18, supplément 2, September 2016

Illustrations
In 2011, Corbett et al. reported six patients with a progressive myoclonus epilepsy (PME) (Minassian et al., 2016) phenotype whose main cardinal features were onset of ataxia in the first years of life, appearance of action myoclonus and seizures later in childhood, and loss of independent ambulation in the second decade. Cognition was not typically affected, although mild memory difficulties occurred for some in the third decade. This condition was found to be associated with a homozygous mutation in GOSR2 (c.430G>T, p.Gly144Trp), a Golgi vesicle transport gene. This p.Gly144Trp mutation gives rise to a loss of function and results in failure of the GOSR2 protein to localize to the cis-Golgi. Following this, the clinical and neurophysiological characteristics of PME associated with GOSR2 mutation were further detailed in 12 patients (including the original six patients described by Corbett et al. [2011]); all patients had the same homozygous mutation (c.430G>T, p.Gly144Trp) (Boissé Lomax et al., 2013). Interestingly, the birthplaces of all these patients (including the birthplaces of the ancestors of one Australian patient) clustered around the North Sea (hence the eponym for this type of PME of ‘North Sea progressive myoclonus epilepsy’ by Boissé Lomax et al. [2013]). This geographic distribution suggests that the identified GOSR2 mutation may have spread along the North Sea at the time of the Viking conquests in the
Gene identification
The GOSR2 gene was identified as a causative gene for PME through the genetic and molecular analyses of a family with one affected child. The proband was an Australian female, born to second-cousin parents of British origin. She, her unaffected brother, and her parents were genotyped using Affymetrix 250K Nsp SNP chips for genome-wide linkage mapping. Homozygosity mapping was performed and a single suggestive linkage region on chromosome 17 was identified, with a maximum possible LOD score of 1.93. Sequence capture followed by next-generation sequencing was carried out and a homozygous variant in GOSR2 (MIM: 604027), c.430G>T, p.Gly144Trp, (NM_004287.3), was identified as the possible causative mutation. Based on a sequence analysis of a cohort of 73 unrelated individuals with molecularly unsolved PME, five additional individuals (from four families) were identified who were homozygous for the same GOSR2 variant, c.430G>T, p.Gly144Trp (Corbett et al., 2011). No consanguinity was reported in any of the families. The c.430G>T GOSR2 variant was not found in 584 chromosomes from unaffected individuals or in dbSNP132, leading to confirmation that this was the causative mutation. Of the four additional families, one was of German ancestry and three were Dutch. Further analysis of one affected individual from each family with both microsatellite markers and Illumina 610 quad SNP chips revealed a founder mutation that was most likely to be of European ancestry (Corbett et al., 2011).
GOSR2 is a member of the Qb-SNARE family of vesicle docking proteins. There are three known alternatively spliced isoforms of GOSR2 and all are predicted to be affected by the c.430G>T, p.Gly144Trp mutation. The mutated glycine 144 residue (G144) is within the Qb-SNARE domain of the GOSR2 protein and shows high evolutionary conservation from mammals to yeast. Initial investigations into the pathogenic mechanisms due to the mutation have shown that the GOSR2 p.Gly144Trp mutant protein fails to localize to the cis-Golgi (Corbett et al., 2011).
Clinical features associated with GOSR2 mutation
The distinct clinical features of PME associated with GOSR2 mutations are early-onset ataxia (at around 2 years of age), onset of action myoclonus and seizures at around 6 years of age, and scoliosis in adolescence and the absence of cognitive deterioration during evolution, although mild cognitive decline may be observed in the later stages. The clinical history of Case 1 in the original report by Corbett et al. (2011) is paradigmatic in terms of illustrating the characteristics and course of the disease. This patient began to have difficulty of walking and was found to be areflexic at the age of 2, but development was otherwise normal. At the age of 7, she had a tremor, and it became clear that action myoclonus and occasional absence seizures were present. At the age of 13, she began having drop attacks as well as major convulsive seizures. The patient required a wheelchair from the age of 14 due to falling attacks and was unable to walk unaided from the age of 16. Notably, the patient had severe scoliosis which required surgical correction. By the age of 22, the patient was confined to bed and she died aged 32 as the result of complications associated with uncontrolled myoclonus. The patient's intellect was preserved until the latter few years of her life, when there was mild cognitive impairment. Autopsy showed mild cerebral atrophy and a lack of gross structural abnormalities. Histological examination revealed subtle, Alzheimer type II gliosis in the basal ganglia region (consistent with metabolic derangement related to her agonal state) and minor loss of Purkinje cells and gliosis in the cerebellar vermis.
The clinical features and evolution of PME caused by mutation of GOSR2 were detailed in the study by Boissé Lomax et al. (2013) who described the original 6 patients with the homozygous GOSR2 c.430G>T, Gly144Trp mutation (Corbett et al., 2011), as well as six new patients (from 11 families) who were molecularly identified with the same mutation. The clinical presentation in the 12 patients was remarkably similar, with features of early-onset ataxia (on average at 2 years of age), followed by myoclonic seizures at the average age of 6.5 years. During the course of the disease, all patients exhibited multiple seizure types, including generalized tonic-clonic seizures, absence seizures, and drop attacks. The patients also uniformly displayed highly photosensitive generalized myoclonus that worsened with action or emotional stress, but was minimal at rest and almost completely absent during relaxation. In some patients, myoclonus and drop attacks were made worse by fever. One patient presented with periods of ‘status myoclonicus’, characterized by continuous myoclonus lasting for hours up to a day at a time. All patients developed scoliosis by the time they reached adolescence, making this an important diagnostic feature. Additional skeletal deformities were present, including pes cavus in 4 patients and syndactyly in two patients. Notably, cognition was preserved in the context of severe motor disability until the later stages of the disease. The progression of the disease showed a relentless decline; patients became wheelchair-bound (at a mean age of 13 years) and four died during their third or fourth decade of life.
An additional patient with PME caused by mutation in GOSR2 has been reported by Praschberger et al. (2015). This patient was a 61-year-old female presenting with a PME phenotype and was found to be compound heterozygous for two GOSR2 mutations; the known c.430G>T, Gly144Trp mutation and a novel c.491_493delAGA (p.Lys164del) mutation. She presented with mild ataxia at the age of 2 years, as well as transient episodes of motor deterioration triggered by infection and fever. At the age of 14 years, she started to suffer from action myoclonus and seizures. In her thirties, she was wheelchair-bound. Scoliosis and areflexia were also noted. No cognitive deterioration was observed throughout the course of the disease, although mild cognitive decline was detected after repeated neuropsychological testing. The most relevant distinctive clinical feature of this patient was the milder course of the disease, at variance with the previously reported patients with PME associated with the homozygous GOSR2 Gly144Trp mutation.
Finally, van Egmond et al. (2014) reported the same homozygous c.430G>T, p.Gly144Trp GOSR2 mutation in five Dutch patients with progressive myoclonus ataxia (PMA) (also known as Ramsay Hunt syndrome). These patients showed clinical features that were very similar to those previously described for GOSR2 mutation-positive PME patients. However, differences include the fact that in the five patients with PMA, cognitive function was not seen to decline (however, only one patient was in his third decade while the others were younger) and scoliosis was observed in only three of the five subjects.
Neurophysiological investigations
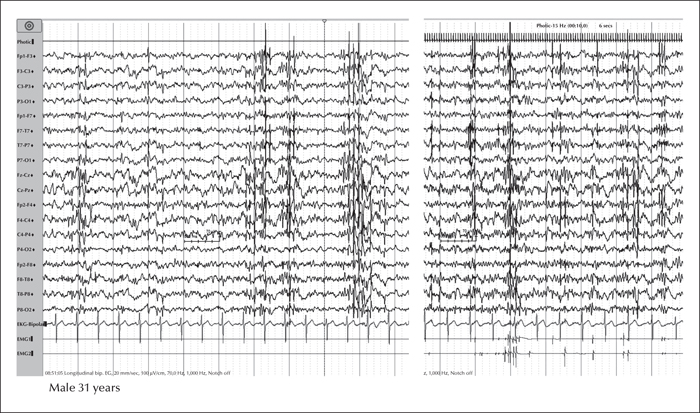
EEG analysis of GOSR2 mutation-positive PME patients reveals generalized spike-and-slow-wave discharges with a posterior predominance, often with a slow background. The generalized discharges are highly photosensitive. Focal or multifocal discharges can also be observed (Boissé Lomax et al., 2013) (figure 1). Nerve conduction studies have been reported to be consistent with a mild, predominantly axonal peripheral neuropathy, while electromyography was normal in all patients with the exception of one in the series reported by Boissé Lomax et al. (2013). Signs of sensory neuronopathy, anterior horn cell involvement, or both, were detected in all patients with absent reflexes reported by van Egmond et al. (2014).
Multimodal evoked potentials were shown to be unremarkable (Boissé Lomax et al., 2013).
Brain magnetic resonance imaging studies have displayed essentially normal findings or generalized cerebral and cerebellar atrophy (Boissé Lomax et al., 2013; Prachschberger et al., 2015). Elevation of serum creatine kinase levels (median: 734 IU), in the context of normal muscle biopsies, was reported for all patients in the study of Boissé Lomax et al. (2013), but was not a uniform feature in the series described by van Egmond et al. (2014).
Conclusions
GOSR2-associated PME has a relatively homogeneous clinical presentation, characterized by a pattern of early-onset ataxia, areflexia, action myoclonus and seizures, scoliosis, elevated creatine kinase levels, relative preservation of cognitive function until the late stages of the disease, and relentless disease course. Thus far, the same homozygous c.430G>T (p.Gly144Trp) mutation has been detected in all reported patients with GOSR2-mediated PME, with the exception of one patient with a milder disease course who was heterozygous for the known c.430G>T (p.Gly144Trp) mutation and a novel c.491_493delAGA (p.Lys164del) GOSR2 mutation. The identification of additional patients will contribute to further expanding the phenotype and genotype and will add to our knowledge of GOSR2-related disease.
Disclosures
None of the authors have any conflict of interest to disclose.
The present manuscript is part of a Epileptic Disorders/Mariani Foundation supplement on Progressive Myoclonus Epilepsies, downloadable as a whole by visiting www.epilepticdisorders.com.