Epileptic Disorders
MENUEpileptic encephalopathy with microcephaly in a patient with asparagine synthetase deficiency: a video-EEG report Volume 21, numéro 5, October 2019

Asparagine synthetase deficiency (ASNSD, MIM # 615574) is a rare autosomal recessive neurometabolic disorder caused by mutations in the asparagine synthetase (ASNS; MIM * 108370) gene.
It was first described in 2013 in four families with homozygosity or compound heterozygosity for missense mutations in ASNS (Ruzzo et al., 2013).
The severe neonatal infantile phenotype is characterized by congenital and progressive microcephaly, jitteriness, erratic myoclonus, spasticity, epilepsy, severe developmental delay, decreased cerebral volume and size of pons, gyral simplification, very thin corpus callosum, and delayed myelination. Other frequently reported signs are relapsing vomiting, feeding difficulties, and cortical blindness.
After the first description, reports of other patients affected by various mutations in the ASNS gene (Ruzzo et al., 2013; Alfadhel et al., 2015; Ben-Salem et al., 2015; Palmer et al., 2015; Gataullina et al., 2016; Seidahmed et al., 2016; Gupta et al., 2017; Sacharow et al., 2017; Sun et al., 2017; Yamamoto et al., 2017; Abhyankar et al., 2018; Galada et al., 2018; Schleinitz et al., 2018) led to increased knowledge of the phenotype, thus allowing suspicion of the disease based on clinical features.
However, the expression of the disease can vary, and only in a few cases with early clinical suspicion, in which biochemical findings support the diagnosis, is the ASNS gene screened by Sanger sequencing. In general, whole-exome sequencing (WES) remains the most powerful and the only reliable diagnostic method. Nevertheless, often the whole diagnostic process takes a long time for a disease in which early genetic counselling is fundamental.
Therefore, we strongly believe that a detailed description of the epilepsy phenotype with extensive electroclinical data, as reported here, might facilitate early diagnostic suspicion, leading to molecular genetic investigation, as already proposed in one previous publication (Gataullina et al., 2016).
Case study
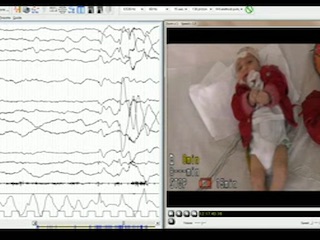
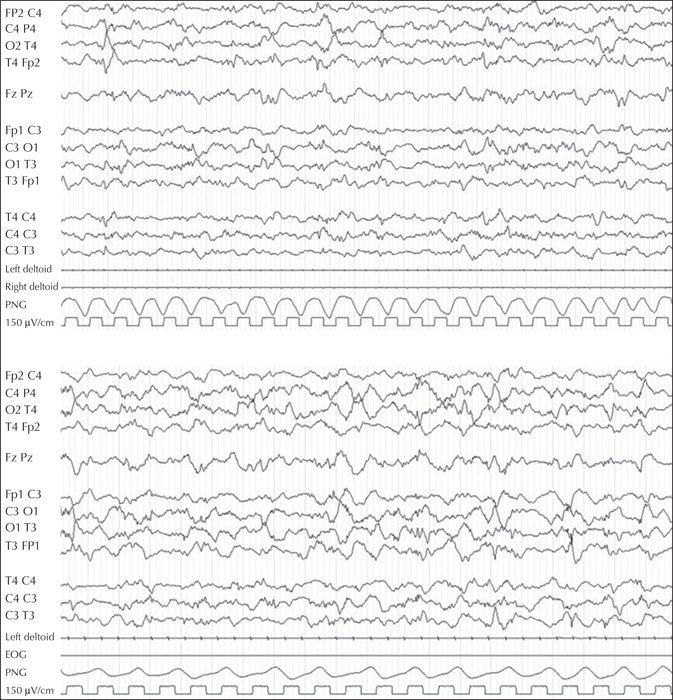
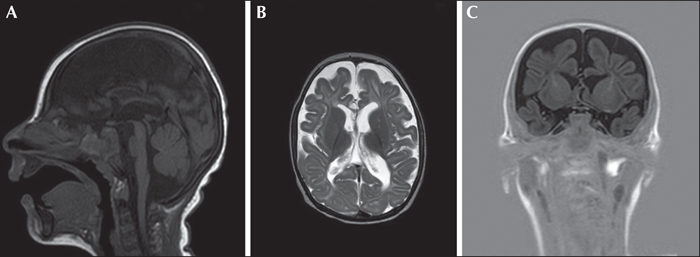
A first male child was born at 39 weeks and two days of gestational age from healthy parents, following an uncomplicated pregnancy. His birth weight and length were normal, head circumference was 34.1 cm (1 SD below the mean), and APGAR score was 9-10. From the second day, he had provoked diffuse myoclonic jerks, with normal glucose and calcium levels. At 53 days of life, the child had irritability, jitteriness, feeding difficulties with swallowing dysfunction, frequent vomiting, and a downward deflection of the growth curve. On admission to the paediatric department of his district hospital, microcephaly was evident, and head control and eye contact were absent. Serology (CMV, toxoplasma, rubella) was negative. Amino acid chromatography based on plasma and liquor were not performed at that time. Organic acid chromatography was normal. The MRI showed brain atrophy with reduced myelin and simplified gyration. Visual evoked potentials were normal. The EEG did not show epileptic anomalies. At three months of age, when we observed the child for the first time, he presented with microcephaly, cortical blindness, and marked axial hypotonia with limb spasticity. Gastroesophageal reflux disease, frequent vomiting, swallowing dysfunction, gastroesophageal incoordination, and profound hyperexcitability with excessive startle reflex persisted. Focal seizures, mostly with clonic facial and upper limb jerks in clusters, and non-epileptic and epileptic myoclonic jerks, were observed. The EEG showed poor background activity, focal posterior and generalized ictal activity during wakefulness, and multifocal spikes in sleep (figure 1). Epileptic spasms were registered during wakefulness (see video sequence). Plasma amino acid chromatography showed an asparagine level of 45 μmol/L (20-90), aspartic acid level of 0 (0-20), and glutamine level of 1,654 (246-1,180). Urinary amino acid chromatography showed a level of asparagine at 121 μmol/L (252-1,280) and glutamine at 3,736 (670-1,562). Carbohydrate deficient transferrin was normal. Lactic acid and glucose in CSF were normal. At 10 months, brain MRI confirmed decreased cerebral volume and the size of the pons, gyral simplification, and a very thin corpus callosum, as well as delayed myelination (figure 2). Seizures were resistant to clonazepam, ACTH, and vigabatrin, whereas a better response was observed with topiramate and valproic acid. A pyridoxine challenge was never performed given the rapid progression of clinical and neuroradiological features.
Genetic study
High-density SNP-array analysis did not reveal microdeletions or duplications. Due to remote parental consanguinity, runs of homozygosity (ROH) were investigated, leading to the detection of three large ROH. Further investigation of these regions for autosomal recessive OMIM genes led to identification of the ASNS gene.
Asparagine synthetase deficiency disease closely matched the phenotype of the patient, leading to Sanger sequencing analysis of the ASNS (NM 183356) gene, which revealed a homozygous novel variant, c.761G>T (p.Gly254Val). The detected variant is predicted to be deleterious based on all in silico prediction webtools used (MutationTaster, PolyPhen2, SIFT, Provean) and the parental segregation analysis confirmed the heterozygous state.
Discussion
ASNSD together with glutamine synthetase deficiency and serine biosynthetic disorder is included in the recently recognized group of conditions resulting from the inability to synthesize a non-essential amino acid.
ASNSD is caused by homozygous or compound heterozygous mutation in the ASNS gene, which encodes the asparagine synthetase protein. It is involved in the synthesis of amino acid asparagine by transfer of ammonia from glutamine and aspartate. It has been proposed that insufficient supply of asparagine within the brain underlies brain malformation and malfunction (Ruzzo et al., 2013). In fact, proliferation of fibroblasts derived from patients with ASNSD was markedly reduced under conditions of asparagine deprivation (Palmer et al., 2015).
The pathophysiology of ASNSD is probably more complex than simple CNS asparagine deficiency and it is still unclear whether the whole phenotypic spectrum can be attributed to the sole perturbation in asparagine metabolism. It has been hypothesized that balance dysregulation of the involved amino acids, with accumulation of aspartate/glutamate, could result in enhanced excitability (Lomelino et al., 2017; Sacharow et al., 2017).
Epilepsy is considered not specific and myoclonic, tonic, spasm and generalized tonic-clonic seizures, which are refractory to antiepileptic medications, are reported (Gupta et al., 2017). EEG shows severe and diffuse brain dysfunction with overall slowing and flattening of amplitude (Schleinitz et al., 2018), multiple independent spike foci, burst suppression, hypsarrhythmia, and disorganized background (Gupta et al., 2017).
Gataullina et al. (2016) described two patients with a particular electroclinical pattern, distinct from both neonatal myoclonic encephalopathy (with suppression-burst and myoclonus preceding discontinuous tracing by several weeks) and West syndrome, although one sibling exhibited spasms and modified hypsarrhythmia. In the authors’ opinion, the pattern is reminiscent of neonatal pyridoxine-dependent epilepsy due to PNPO or ADLH7A1 mutation.
The electroclinical phenotype of our case confirms Gataullina's observation. In particular, in our patient, excessive irritability, jitteriness, and feeding difficulties with oromotor incoordination were present from the first days of life. A first EEG did not show epileptic abnormalities, however, the absence of a polygraphic recording did not allow better definition of the nature of the movement disorder. At three months of life, the child showed, at the same time, multiple seizure types (predominantly focal clonic with facial grimacing, and myoclonic seizures) intermingled with jitteriness and myoclonic non-epileptic jerks, as a probable expression of a widespread enhanced excitability, however, the lack of amino acids in the liquor did not support this hypothesis.
The EEG showed paroxysmal multifocal and generalized anomalies. Low-amplitude activity, disorganized background, as well as a period of attenuation were also observed, coherently with the “progressive” nature of the disease. A classic hypsarrhythmic pattern did not appear even when epileptic spasms were evident.
Recently, Dulac (2018) included the epileptic phenotype of ASNS in the group of early progressive myoclonic epilepsies, symptomatic of deficiency of non-essential amino acid synthesis (serine and glutamine) and of pyridoxine/pyridoxal-phosphate dependency. Unlike these deficient metabolic diseases, there is no effective treatment for ASNSD. The presence of congenital microcephaly and early postnatal presentation suggest that supplementation with asparagine would be beneficial only if started prenatally (Ruzzo et al., 2013). Moreover, asparagine supplementation can worsen the clinical picture. In fact, asparagine uptake across the blood brain barrier is the effect of a facilitative transport system, and artificially elevating blood asparagine levels too high may inhibit uptake of other amino acids due to competition for shared transporters (Lomelino et al., 2017). Interestingly, in our patient, a better response for seizure control and global hyperexcitability was observed on valproic acid, similarly to the patient described by Alrifai and Alfadhel (2016). The authors hypothesized a specific efficacy of the drug due to its proposed mechanism of action, namely the selective decrease in aspartate release at excitatory nerve terminals.
Epilepsies of genetic origin frequently display age-dependent electroclinical features, presumably related to the sequential development of excitatory and inhibitory pathways in the neonatal brain. The pathophysiology of seizures is probably complex in ASNSD, involving multiple, interrelated mechanisms, also, the neuropathological findings may account for several epileptic features (Gataullina et al., 2016)
Knowledge on the extent of clinical and EEG characteristics of ASNSD is rapidly expanding due to the availability of WES. WES remains the most powerful and the only reliable diagnostic method for ASNSD and many other encephalopathies with early intractable epilepsy, severe developmental delay, and microcephaly. Nevertheless, some specific reported features, such as a neurodevelopmental isolated phenotype with early-onset severe feeding difficulties, cortical blindness, rapidly progressive microcephaly with an enlarged ventricular system and pericerebral spaces, reduced myelination, a simplified gyral pattern on MRI, a movement disorder with severe hypertonic dyskinetic quadriplegia, and signs of hyperexcitability including hyperekplexia, may lead to a suspicion of the disease. Moreover, the coexistence of an electroclinical phenotype characterized by an early onset of epilepsy with multiple types of intractable seizures and low-amplitude disorganized EEG with multifocal paroxysmal activity, evolving into a discontinuous pattern, can be considered an important diagnostic clue and guide the diagnostic work-up, in some cases leading to Sanger sequencing. Beside the clinical picture, in our case, careful family history taking was essential, leading to investigation of ROH, based on SNP-array analysis, and identification of ASNS gene involvement and mutation analysis based on Sanger sequencing.
Disclosures
None of the authors have any conflict of interest to declare.
* The case was presented at the last “Riunione Policentrica, Lega Italiana Contro l’Epilessia” (January 2018).