Epileptic Disorders
MENUDevelopmental pharmacology of benzodiazepines under normal and pathological conditions Volume 16, numéro spécial 1, October 2014

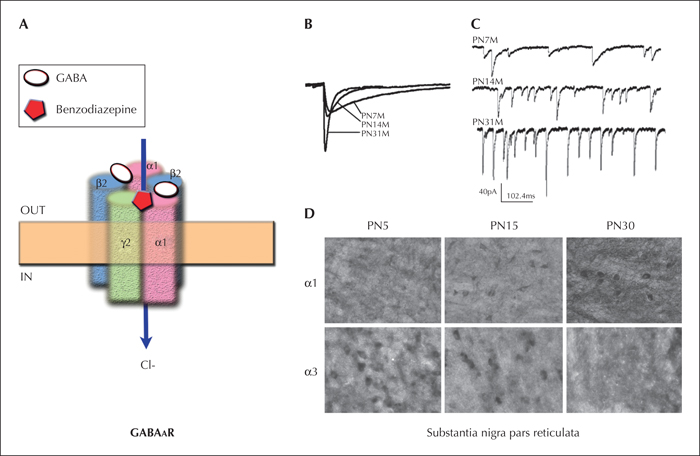
Benzodiazepines are allosteric agonists of GABAA receptors (GABAARs), pentameric ligand-activated Cl- channels that typically mediate Cl- inflow leading to neuronal hyperpolarisation and fast inhibitory postsynaptic currents. Benzodiazepines act in the presence of GABA, and their effects greatly depend upon the type of subunits present in the GABAARs. Their affinity is greatest for GABAARs containing α1 and γ2 subunits. The inhibitory effects of benzodiazepines in combination with their availability as formulations that permit rapid and flexible delivery (e.g. buccal, nasal, rectal), even in situations when intravenous access is not available, have established them as the first-line rescue drugs or treatment for rapid cessation of ongoing seizures throughout life.
However, the effects of GABAAR agonists may change under certain normal or pathological situations, in which either the subunit composition is not optimal or the function of GABAARs is altered. Here, we will review the animal studies that have highlighted the developmental changes in GABAAR physiology and pharmacology of their agonists, including benzodiazepines, and discuss these findings with regards to their potential relevance for the clinical use of benzodiazepines in the treatment of ongoing seizures, and especially in very young individuals.
GABAAR structure and pharmacology
There are currently 16 known subunits in mammals (six α, three β, three γ, and one δ, ε, θ, and π). Each GABAAR consists typically of five subunits, with the most commonly observed arrangement being a combination of two α, two β, and one γ subunit (figure 1). Subunit composition dictates a number of receptor characteristics, including localisation within each cell or in various brain regions, affinity for ligands and drugs, as well as regulation by specific signalling pathways. For example, typically (but not always), GABAARs that contain γ subunits are located post-synaptically, and their activation is recorded as phasic inhibitory postsynaptic currents (IPSCs) (Mody and Pearce, 2004; Farrant and Nusser, 2005; Mohler, 2006a). However, substitution of a γ for a δ subunit causes extrasynaptic localisation and results in tonic GABAAR activation by ambient GABA molecules (tonic currents) (Nusser et al., 1998; Farrant and Nusser, 2005). Specific subunit composition is not only responsible for subcellular localisation, but also impacts the kinetics of the receptors, as well as their affinity for certain ligands (Mohler, 2006a). As an example, α3 subunits located in extrasynaptic receptors have been shown to desensitize much more slowly than α2 subunits located in synaptic receptors (Devor et al., 2001).
Whereas the GABA binding site lies between an α and a β subunit, the benzodiazepine binding pocket is between an α and a γ subunit, and their affinity is greatest for GABAARs which contain α1 and γ2 subunits, intermediate for α2 or α3-containing GABAARs, low for δ-containing GABAARs, and absent for those that contain α4 or α6 subunits. Consequently, the primary target of benzodiazepines is the potentiation of postsynaptic GABAARs, that are already activated by synaptically released GABA quanta, and which mediate phasic GABA inhibition.
Since specific subunit composition has such far-reaching effects on kinetics and pharmacology, the differential expression of GABAAR subunits across brain regions, cell types, between genders, and as the brain develops, adds to the diversity of their final effects (Mohler, 2006a; Mohler, 2006b). The regional differences in subunit expression are also thought to contribute to the different functional effects of agonists that prefer certain subunit combinations (Mohler, 2006a). For example, α1 is found more in the thalamus, cortex, brainstem, hypothalamus, and hippocampus, and agonists may have antiseizure, sedative, and amnesic effects. In contrast, selective agonists for α2 and α3-containing GABAARs exhibit anxiolytic or myorelaxant effects. However, most of these studies are largely on adult populations and as we will discuss later, the effects in the developing brain may be quite different.
GABAAR structure and pharmacology change through development
During normal development, the affinity of GABAARs for agonists or antagonists changes due to age-, region- and sex-specific patterns of GABAAR subunit expression, subcellular localisation, or mRNA editing. Most of these studies have been performed in rodents. The significantly shorter lifespan of rodents (approximately two years) warns that brain development in rodents occurs relatively faster than in humans. In general, a postnatal day (PN) 8-13 rat or mouse is considered to be equivalent to a human newborn full-term baby, based on gross measures of brain growth (weight, protein/DNA content). However, unlike human babies, eye opening only occurs between PN13-15 in rodents. Weaning from the dam usually takes place at around PN21 and puberty starts between PN32-35, whereas adulthood begins on PN60. In fact, when comparing rodent to human development, each developmental process may mature at its own tempo and cross-species extrapolation may not be very accurate.
GABAAR subunit expression changes through development following cell-type and region-specific patterns. Although such developmental changes are widespread, there is considerable diversity across regions and cell types. However, a well-documented “subunit switch” between the α2/α3 and α1 subunits has attracted a lot of interest as it has been observed in multiple studies, regions, and species, and may potentially account for important age-related differences in GABAAR inhibitory responses and their pharmacology. Laurie et al. described increasing α1 subunit expression with age in the telecenphalic cortex, diencephalon, thalamus, hippocampus, and cerebellum (Laurie et al., 1992). At the same time, a simultaneous decline in α3 (telencephalon, cortex, diencephalon, thalamus, and cerebellum) and, in certain regions, α2 (diencephalon, thalamus) occurs. Similar observations have also been documented in the basolateral amygdala (Ehrlich et al., 2013) and substantia nigra pars reticulata (SNR) (Chudomel et al., 2009) (figure 1). The replacement of α2/α3 subunits by α1 alters not only the kinetics of phasic GABAAR inhibitory postsynaptic currents (IPSCs), rendering them faster, but also decreases the affinity of these receptors to drugs such as zolpidem or benzodiazepines. For example, in the rat SNR, PN5-9 SNR GABAergic neurons had low frequency and slow GABAAR-IPSCs, whereas in peripubertal PN28-32 rats, IPSCs were recorded with high frequencies and rapid kinetics and were more sensitive to zolpidem, a preferential agonist of α1 (Chudomel et al., 2009). A similar switch has also been documented in the monkey prefrontal cortex, with high levels of α2 mRNA in one-week-old monkeys and a gradual replacement by high α1 mRNA levels in adult monkeys (Hashimoto et al., 2009). Studies in humans have been limited to post-mortem tissues, for obvious ethical reasons. Two independent studies in the human cerebral cortex reported increasing expression of α1 and γ2 mRNA between the neonatal and adult ages, while the level of mRNA of other subunits, such as α2 (Fillman et al., 2010) or α4 (Jansen et al., 2010), declined.
Another point of developmental regulation is at the mRNA editing level, which may change the function of the translated proteins. In the case of α3 subunit, the level of RNA editing leading to an isoleucine-to-methionine substitution in the region coding the third transmembrane domain was very low during mouse embryonic development, reached maximal levels at PN7, and was observed in 90% of adult mouse brains (Rula et al., 2008). The edited and non-edited forms of α3 subunit confer distinct channel properties. GABAARs containing non-edited α3 activate rapidly and deactivate more slowly than those that contain edited α3 subunits, but also exhibit stronger outward rectification (i.e. Cl- influx). This particular feature renders the non-edited α3 subunit-containing GABAARs more capable of mediating shunt inhibition in the face of depolarising currents, and can therefore be protective early in development, when GABAAR currents can be depolarising, as will be discussed in the following section.
Changes in depolarising to hyperpolarising GABAAR signalling with age
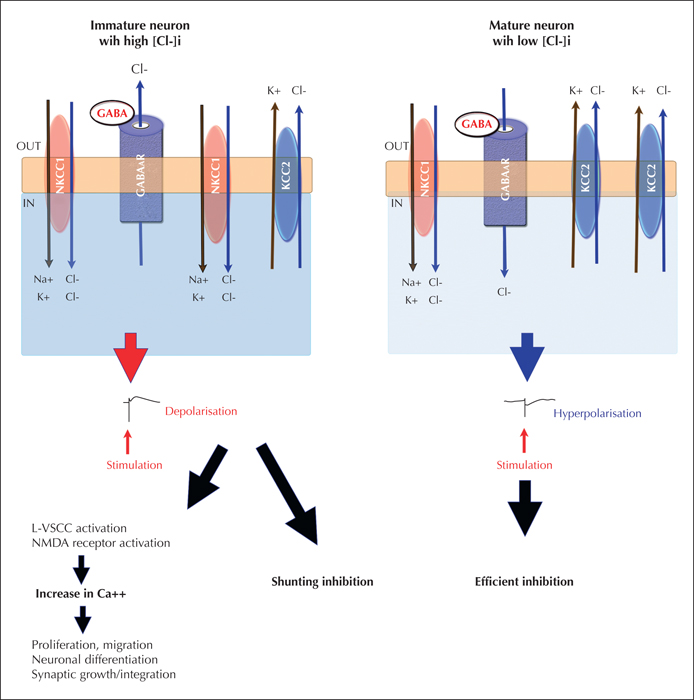
The direction of GABAAR responses depends upon the Cl- gradient between the intra- and extracellular space and also, to a degree, on HCO3-. The intracellular Cl- concentration is controlled by cation-Cl- cotransporters, of which the most widely studied is the Cl- importer Na+/K+/Cl- cotransporter 1 (NKCC1) and the Cl- exporter K+/Cl+ cotransporter (KCC2) (figure 2). Early in life, in most studied neurons, there is a relatively high intracellular Cl- concentration, due to a relative abundance of NKCC1 over KCC2. As a result, opening of the GABAAR mediates Cl- efflux, leading to depolarisation. The early immature depolarising effects of GABAA receptors are important for development; nevertheless, they lead to calcium processes necessary for neuronal processes including cell migration and differentiation (Ben-Ari, 2002; Galanopoulou, 2008a). Precocious deprivation of depolarising GABA from developing neurons may lead to dysmature features, such as underdeveloped arborization of neuronal processes in cortical neurons (Cancedda et al., 2007; Wang and Kriegstein, 2008; Wang and Kriegstein, 2011). It should be noted that these normal depolarising effects of GABA are not necessarily excitatory, since they may not always trigger action potentials. Furthermore, in the setting of excessive excitation, as during seizure activity, GABAAR agonists may still exert inhibitory effects, through shunting inhibition, although this type of inhibition is less effective (Staley and Mody, 1992).
During development there is a gradual shift, from an NKCC1 dominant state, early on, to a KCC2 dominant state in more mature neurons. This eventually reduces the intracellular Cl-, resetting the Cl- electrochemical reversal potential, and resulting in Cl- influx and hyperpolarisation after GABAAR activation (Plotkin et al., 1997; Rivera et al., 1999) (figure 2). The timing of shift from depolarising to hyperpolarising GABAAR signalling varies across brain regions, neuronal types, and between sexes, adding another level of complexity to the understanding of GABAergic signalling. Such examples are the substantia nigra pars reticulata (SNR) and CA1 pyramidal neurons of the hippocampus of Sprague-Dawley rats, where GABAAR activation elicits sex-specific effects, with delayed disappearance of depolarising responses in males compared to females (Galanopoulou et al., 2003; Kyrozis et al., 2006; Galanopoulou, 2008b). Neurons in the SNR of male rats were shown to start manifesting hyperpolarising GABAAR responses by postnatal day (PN) 17, while in females the switch occurred much earlier, at PN10 (Kyrozis et al., 2006), consistent with increased KCC2 mRNA in the SNR of female rats at all ages (Galanopoulou et al., 2003). This sexually dimorphic pattern continues across other brain regions, including CA1 pyramidal neurons. Again, female rats showed isoelectric or mildly hyperpolarised GABAAR as early as PN4, while males started to have hyperpolarising responses around PN14 (Galanopoulou, 2008b). Systemic administration of GABAAR agonists have also demonstrated sex-specific effects in intracellular processes, such as phosphorylation of the cAMP responsive element binding protein (pCREB), which are probably linked to the depolarisation-induced calcium rises (Auger et al., 2001; Galanopoulou et al., 2003; Perrot-Sinal et al., 2003; Galanopoulou, 2006; Mantelas et al., 2007; Galanopoulou, 2008c). Sex differences in KCC2 and/or NKCC1 have been proposed to underlie the earlier onset of hyperpolarising GABAAR signalling in females (Galanopoulou et al., 2003; Galanopoulou, 2008b; Damborsky and Winzer-Serhan, 2012; Murguia-Castillo et al., 2013). KCC2 expression is also under the control of gonadal hormones in immature rat SNR neurons. According to a proposed hypothesis, the high endogenous levels of oestrogenic derivatives of testosterone aromatization in the newborn male rat brain may actually delay the developmental rise of KCC2 and the appearance of hyperpolarising GABA in certain brain regions, such as the SNR (Galanopoulou, 2008c).
In humans, the only evidence corroborating the idea that there may be depolarising GABA in very immature neurons comes from post-mortem studies, where high NKCC1 and low KCC2 expression were shown in cortical extracts from neonates, followed by a rapid decline in NKCC1 and gradual increase in KCC2 during the first few months of life (Dzhala et al., 2005; Jansen et al., 2010).
GABAAR agonists have age-specific effects
What is the impact of all these structural and functional changes of GABAARs upon the effects of drugs such as benzodiazepines or barbiturates through development? Animal studies have addressed this question in normal animals and have supported the idea of age-specific effects of GABAergic drugs. Increased apoptotic death and reduced neurogenesis is observed in one-week-old rats exposed to benzodiazepines or phenobarbital (Bittigau et al., 2002; Bittigau et al., 2003; Stefovska et al., 2008). Similar findings were observed with other antiseizure drugs, suggesting that the immature brain may be more dependent upon a higher level of neuronal activity than the adult for survival. Midazolam is reported to decrease the mechanical reflex threshold and increase the magnitude of mechanical and thermal reflexes in neonatal rats, but had no effect in juvenile rats (Koch et al., 2008). In the same study, midazolam had sedative effects in PN10 and PN21 rats but not in PN3 rats. Although it is tempting to associate these paradoxical effects of midazolam in newborn rodents with the early depolarising effects of GABAAR signalling, the answer is not always as simple. The final output of the inhibition or activation of a neuronal cluster in the brain is largely dependent on the way a neuronal network is organised, interconnected, and functionally active. For example, infusions of the GABAAR agonist muscimol in the SNR, which although ultimately silences local neuronal activity, may produce anticonvulsant or proconvulsant effects, depending on the age of the rat, whether it is male or female, and whether it is infused in the anterior or posterior SNR (Veliskova and Moshé, 2001; Veliskova and Moshé, 2006).
Even more importantly, drugs are typically given to target a specific disease process or symptom and their effects cannot be extrapolated to those observed in a diseased brain. For example, although benzodiazepines may potentially accentuate cell death in newborn rats, these studies were performed in normal rodents and not in animals that exhibit status epilepticus, which, on its own merit, may have adverse developmental outcomes. Therefore, careful evaluation, including evaluation of long-term outcomes, is warranted when assessing the efficacy and tolerability of these drugs in very young subjects.
Seizures and epilepsy can disrupt GABAAR signalling
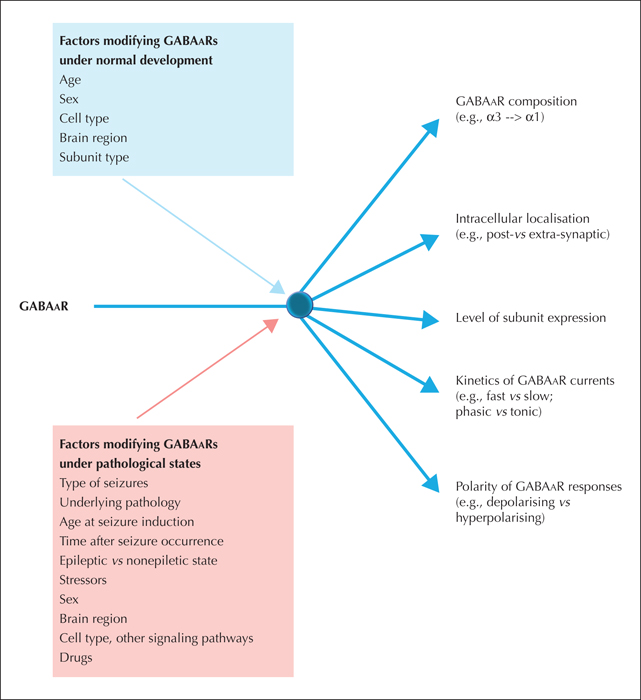
Multiple studies have investigated the association between either induced seizures, the epileptic state or different aetiology and the expression and function of GABAAR subunits (reviewed in Galanopoulou and Moshé [2014]). Using the kainic acid or the pilocarpine (with or without lithium) models as methods to test the effects of status epilepticus on the expression of GABAAR subunits, there is no singular pattern of changes that emerges. Rather, the effects of seizures depend upon the age at seizure induction, the time-point when subunit expression is studied, and subunit-specific, but also model and region-specific, factors (figure 3). For example, the expression of α1 subunit is increased in the dentate gyrus of adult rats exposed to early-life pilocarpine-induced status epilepticus (Zhang et al., 2004; Raol et al., 2006), regardless of whether these were epileptic or not, but reduced in those exposed to pilocarpine status epilepticus in adulthood (Brooks-Kayal et al., 1998). Expression studies of GABAAR subunits in post-surgical specimens from epilepsy patients with focal cortical dysplasias also suggest a disrupted developmental expression profile of the GABAAR subunits which is also dependent upon the type of dysplasia (Jansen et al., 2010).
A study that has initiated a lot of interest into the potential pathogenic role of depolarising GABAAR signalling in epilepsy reported an aberrant presence of depolarising GABAAR signalling in human epileptic subiculum and paradoxical silencing of interictal epileptic discharges by GABAAR inhibitors (Cohen et al., 2002). This was attributed to reduction in KCC2 expression (Huberfeld et al., 2007). Multiple reports confirmed similar observations in temporal lobe epilepsy (Palma et al., 2006; Munoz et al., 2007) or other pathological epileptic conditions, including hypothalamic hamartomas (Kim et al., 2009), cortical dysplasias (Aronica et al., 2007; Jansen et al., 2010; Shimizu-Okabe et al., 2011; Talos et al., 2012), and tumour-associated epilepsy (Conti et al., 2011). However, based on the human studies, it is not possible to differentiate between the contribution of epilepsy, aetiology, treatments, stressors or other factors on the observed changes in GABAAR signalling. In animal studies, where some of these factors can be better controlled, pathological reversal to depolarising GABAAR signalling with concomitant increase in NKCC1 or decrease in KCC2 have been documented in selected brain regions of adult rodents subjected to status epilepticus (Benini and Avoli, 2006; de Guzman et al., 2006; Pathak et al., 2007; Bragin et al., 2009). On the other hand, the response of immature rats to seizures is again quite different and may be dependent upon sex-specific factors (Galanopoulou, 2008b) or the time after seizure occurrence (Galanopoulou, 2008b; Khirug et al., 2010). For example, kainic acid status epilepticus in neonatal male rats with depolarising GABAAR signalling in the hippocampus causes a premature appearance of hyperpolarising GABAAR signalling, unlike that observed in adult rats (Galanopoulou, 2008b). It is currently, however, unclear whether this is a protective mechanism or if it contributes to the developmental deficits that ensue.
The biggest question therefore is how to use all this knowledge in a manner that will translate into more effective and safer use of GABAergic drugs in humans, particularly in very young individuals, with seizures or epilepsy. Even if we were to account for all the variables involved (age, sex, region, aetiology, cell type, etc.), the expression and function of GABAARs may change with time or in response to external stressors or internal comorbid or physiological factors that will require a live and dynamic predictive modelling method. The discovery and implementation of dynamic, safe biomarkers with sufficient temporo-spatial readout detail will be critical.
Hyperthermic seizures may alter GABAAR signalling
Of particular interest is the role of fever on GABAAR signalling, given the high prevalence of febrile seizures in the paediatric population. Several studies have looked specifically at the role of hyperthermia with variable observations. Hyperthermia can impair GABAAR signalling by decreasing GABAAR IPSCs, increasing GABA uptake, reducing selective α GABAAR subunits, or cause retention of GABAAR subunits in the endoplasmic reticulum (Fujii et al., 2001; Kang et al., 2006; Sharma, 2006; Swijsen et al., 2012). On the other hand, hyperthermia may enhance GABAAR signalling in selective neuronal populations or increase benzodiazepine-sensitive GABAARs in certain brain regions (Gonzalez Ramirez et al., 2007; Swijsen et al., 2012). An interesting study has also proposed that reversal to depolarising GABAAR signalling after hyperthermic seizures induces ectopic granule cells that may contribute to adult epilepsy (Koyama et al., 2012). It should be noted, however, that the role of aberrant seizure-induced neoneurogenesis in epileptogenesis has not been confirmed in other models (Sankar et al., 2000).
Conclusions
In summary, the specific differences in expression and function of GABAARs related to age, anatomical region, sex, receptor subunit, and disease state make it clear that great care should be taken when it comes to using drugs, such as benzodiazepines, particularly early in development. Could more selective α2/α3 GABAAR agonists offer a better efficacy profile for very young patients, given the lower levels of α1 subunit in many brain regions? Could selective enhancers of KCC2 enhance the therapeutic benefit of GABAergic agonists in very young patients? Would methods of tailoring such therapies to deliver them to the specific target areas, for the specific duration of the seizure, or other targeted symptoms prove to be safer? It should be noted, however, that most of the cited studies raising the concern that GABAergic drugs may have adverse effects in the naïve developing brain have been performed in naïve animals. There is no sufficient evidence to indicate that this may also be true for brains that have been exposed to GABAergic drugs with the intent to stop seizures, and it could prove to be challenging to design such a controlled study, given that benzodiazepines have been established as a first-line rescue treatment at all age groups. Yet, as we go forward and more clinical studies are being developed to test the efficacy and tolerability of benzodiazepines as rescue treatments for seizures, it would be worth evaluating not only the immediate efficacy and tolerability but also the longer-lasting consequences of exposure to GABAergic drugs, in both developmental and behavioural outcomes (Galanopoulou, 2008c). The experience in the NEMO (NEonatal seizures using Medication Off-patent) study with the observed ototoxicity from the use of bumetanide, an NKCC1 inhibitor, as adjunctive medication to barbiturates to stop neonatal seizures highlights the concern of safety (Pressler et al., 2013). This is particularly true for regions that are not necessarily within the epileptogenic zone and may become exposed to the drug.
Acknowledgements and disclosures
ASG received research grant support from the NINDS (NS078333), CURE, Autism Speaks, Department of Defense, and the Heffer Family and Siegel Family Foundations, and royalties from Morgan & Claypool Publishers and John Libbey Eurotext Ltd, and consultancy honorarium from Viropharma.
The authors do not have any conflict of interest to disclose in regards to this manuscript.