Epileptic Disorders
MENUDe novo truncating mutation in SCN1A as a cause of febrile seizures plus (FS+) Volume 22, numéro 3, June 2020

Illustrations
- Mots-clés : SCN1A, epilepsy, truncating mutation, febrile seizures plus
- DOI : 10.1684/epd.2020.1167
- Page(s) : 323-6
- Année de parution : 2020
SCN1A is one of the most relevant epilepsy genes. In general, de novo severe mutations, such as truncating mutations, lead to a classic form of Dravet syndrome (DS), while missense mutations are associated with both DS and milder phenotypes within the GEFS+ spectrum, however, these phenotype-genotype correlations are not entirely consistent.
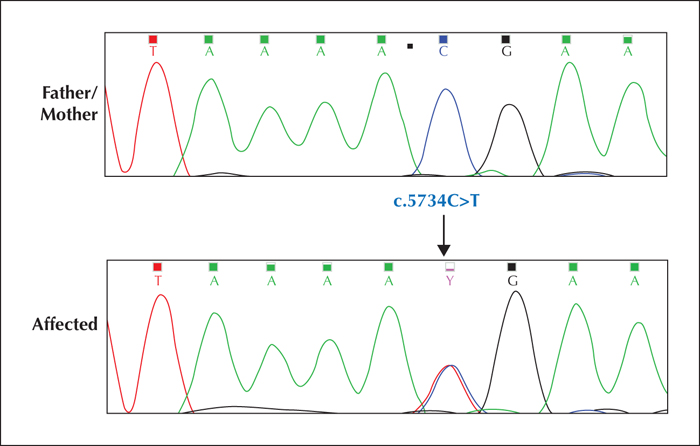
Case report. We report an 18-year-old woman with a history of recurrent febrile generalized tonic-clonic seizures (GTCS) starting at age four months and afebrile asymmetric GTCS and episodes of arrest, suggestive of focal impaired awareness seizures, starting at nine months. Her psychomotor development was normal. Sequencing of SCN1A revealed a heterozygous de novo truncating mutation (c.5734C>T, p.Arg1912X) in exon 26.
Conclusion. Truncating mutations in SCN1A may be associated with milder phenotypes within the GEFS+ spectrum. Accordingly, SCN1A gene testing should be performed as part of the assessment for sporadic patients with mild phenotypes that fit within the GEFS+ spectrum, since the finding of a mutation has diagnostic, therapeutic and genetic counselling implications.