Epileptic Disorders
MENUBiallelic loss-of-function UBA5 mutations in a patient with intractable West syndrome and profound failure to thrive Volume 20, numéro 4, August 2018

- Mots-clés : cerebellar atrophy, epileptic spasm, gene analysis, infantile spasm, ufmylation
- DOI : 10.1684/epd.2018.0981
- Page(s) : 313-8
- Année de parution : 2018
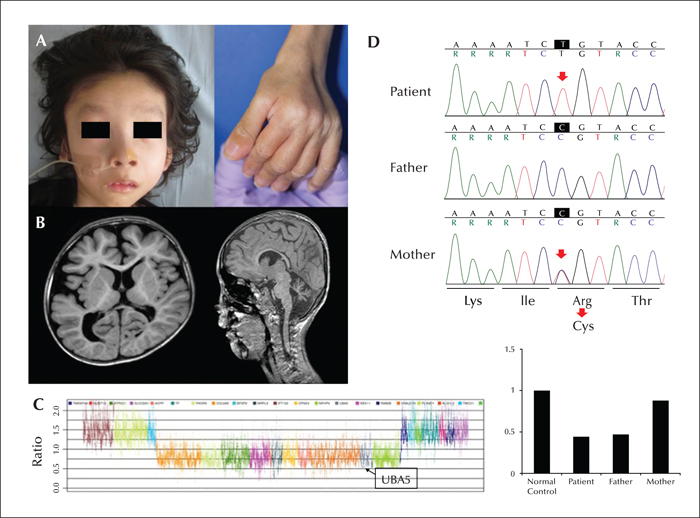
Mutation of the gene encoding ubiquitin-like modifier-activating enzyme 5 (UBA5) causes autosomal recessive early-onset epileptic encephalopathy. UBA5 acts as an E1-activating enzyme in the ubiquitin-fold modifier 1 pathway, which is important for unfolded protein elimination and regulation of apoptosis, and has been linked to human diseases. We identified biallelic mutations in UBA5 in a Japanese boy with intractable West syndrome, profound failure to thrive, and severe cerebral and cerebellar atrophy. The boy presented with epileptic spasms and hypsarrhythmia at the age of three months. He was diagnosed with West syndrome, however, treatments with adrenocorticotropic hormone and several antiepileptic drugs were ineffective. MRI findings were initially normal, but subsequently showed a progression of cerebellar and cerebral atrophy. By the age of seven years, he had not achieved any developmental milestones; he had daily epileptic spasms and tonic seizures and profound failure to thrive. Gene analysis revealed novel compound heterozygous mutations in UBA5; a microdeletion encompassing the entire UBA5 gene and a putative disease-causing missense mutation in the catalytic domain. These biallelic variants may have caused loss of function, accounting for the observed clinical symptoms. Intractable infantile epileptic spasms, failure to thrive, and severe neurological impairment may be characteristic of patients with UBA5 mutations.