Annales de Biologie Clinique
MENURecommandations pour la mise en place et le suivi des contrôles de qualité dans les laboratoires de biologie médicale Volume 77, numéro 5, Septembre-Octobre 2019

Illustrations
Tableaux
-

Tableau 1 -
Tableau 2 -
Tableau 3 -
Tableau 4 -
Tableau 5 -
Tableau 6 -
Tableau 7 -
Tableau 8 -
Tableau 9 -
Tableau 10 -
Tableau 11 -
Tableau 12 -
Tableau 13 -
Tableau 14 -
Tableau 15 -
Tableau 16 -
Tableau 17 -
Tableau 18 -
Tableau 19 -
Tableau 20 -
Tableau 21 -
Tableau 22 -
Tableau 23 -
Tableau 24 -
Tableau 25 -
Tableau 26 -
Tableau 27 -
Tableau 28 -
Tableau 29 -
Tableau 30 -
Tableau 31 -
Tableau 32
Les recommandations formulées dans ce document proviennent essentiellement de la conférence organisée conjointement par LABAC, la SFBC et la FAEEQ à Paris le 30 janvier 2019. Cette journée a permis de débattre des pratiques de contrôle interne de qualité (CIQ), d’évaluation externe de la qualité (EEQ) et d’estimation des incertitudes de mesure (IM) pour des méthodes quantitatives de routine (biochimie, hématocytologie et hémostase). Les analyses de microbiologie (y compris les sérologies infectieuses) sont exclues du champ de ces recommandations, de même que certains examens spécialisés dont l’approche en matière de maîtrise des contrôles de qualité doit être adaptée.
Les différents contributeurs se sont efforcés d’établir des positions explicites qui reflètent la littérature, leurs expériences et leurs connaissances en biologie médicale. Ces recommandations sont à considérer comme une base de réflexion et de travail pour l’ensemble des acteurs de la biologie médicale. Ces recommandations ne prétendent pas répondre à tous les cas de figure envisageables au sein d’un laboratoire. La mise en œuvre d’une stratégie alternative argumentée et objective doit également pouvoir être envisagée. Enfin, ces recommandations sont naturellement susceptibles d’évoluer au fil du temps.
Trois niveaux de recommandations ont été retenus [1] :
- –les pratiques recommandées (réputées conformes aux exigences de la norme NF EN ISO 15189:2012 [2]) : elles proviennent des documents de référence, données faisant l’objet d’un consensus après la lecture des différents articles ou à défaut, reposant sur au moins un article pour lequel la méthodologie et les critères d’interprétation sont solides et robustes (avis d’experts). Elles représentent les « règles de l’art ». Elles sont considérées comme des objectifs de bonne pratique ;
- –les pratiques acceptables : données reposant sur une majorité d’articles ayant fait l’objet d’interprétations variables selon les publications ou, à défaut, un article pour lequel les critères d’interprétation ne sont pas aussi solides que dans la catégorie recommandée (exemple : effectif plus faible, méthodologie statistique utilisée...) ;
- –les pratiques non recommandées (réputées non-conformes aux exigences de la norme NF EN ISO 15189:2012) : données non recommandées faisant l’objet d’un consensus après la lecture des différents articles ou à défaut, reposant sur au moins un article pour lequel la méthodologie et les critères d’interprétation sont solides et robustes (avis d’experts). Les pratiques non recommandées représentent des pratiques qui peuvent remettre en cause la fiabilité des résultats.
Concernant les CIQ, la norme NF EN ISO 15189:2012 [2] définit les exigences suivantes :
- –concevoir des procédures de contrôle permettant de vérifier que la qualité prévue des résultats est atteinte ;
- –utiliser des matériaux de contrôle qui se comportent le plus possible comme des échantillons provenant des patients (notion de commutabilité) ;
- –utiliser régulièrement ces matériaux de contrôle en fonction de la stabilité des méthodes ;
- –évaluer, en cas d’anomalie constatée, l’impact sur les éventuels résultats déjà libérés depuis le dernier CIQ satisfaisant ;
- –revoir les résultats des CIQ régulièrement afin de détecter les tendances.
Le SH REF 02 rev. 05 [3] ajoute les notions de stratégie argumentée et documentée incluant les notions de série, de fréquence de passage, de niveaux utilisés, d’exigences de performances, de règles de validation. Il aborde aussi la conduite à tenir en cas de résultat hors borne du CIQ et l’estimation d’un éventuel impact sur les résultats déjà rendus.
La norme ISO/CEI 17025:2017 [4] introduit la notion suivante : « assurer la validité des résultats ».
L’analyse de risques, la notion de série
Analyse de risques
L’analyse de risques est la première étape essentielle dans la mise en place d’une stratégie de CIQ. Elle consiste en un inventaire des risques analytiques pouvant entraîner un résultat potentiellement erroné (tableau 1).
La liste présentée ci-dessous est non exhaustive et comporte les principaux risques identifiés :
1- Détérioration du réactif au cours de l’expédition
2- Altération de l’étalonnage
3- Présence de micro-caillots obstruant totalement ou partiellement le système de pipetage
4- Maintenances défaillantes ou insuffisantes
5- Détérioration du réactif en cours de stockage dans le laboratoire (défaut de stabilité du réactif) ou péremption du réactif
6- Panne (bloquante et non bloquante) ou dérive du système analytique
7- Conditions environnementales non maîtrisées (température et variation de cette température au cours du temps, humidité, etc.)
8- Dérives au cours du temps de la méthode (dérive et tendance)
9- Erreur par effet opérateur (méthodes manuelles)
10- Erreur par effet opérateur (méthodes automatiques)
Ces risques dépendent de chaque méthode et sont, pour la plupart, connus. Les fournisseurs de dispositifs médicaux de diagnostic in vitro mettent à disposition des moyens de maîtrise. Cependant, ces préconisations peuvent être insuffisantes ou inadaptées et des actions complémentaires sont alors requises. Toutes ces actions devront être associées à des indicateurs qui doivent permettre de suivre la maîtrise des risques en enregistrant les situations pendant lesquelles le risque n’est plus maîtrisé (suivi des non-conformités par exemple). Dans le cadre d’une démarche d’amélioration continue, une réévaluation du risque (avec de nouvelles données d’entrée (non-conformité, audits internes et externes, réclamations, indicateurs, etc.)) est à réaliser avec mise en place d’éventuels nouveaux moyens de maîtrise. Cette démarche « dynamique » permet d’avoir un système pro-actif.
D’autres risques peuvent être identifiés notamment selon la technologie des analyseurs ou les méthodes utilisées (ex. : qualité des consommables, qualité de l’eau, etc.). Il appartient au laboratoire de mettre en place des moyens de maîtrise adaptés.
Les risques maîtrisés par le passage de CIQ sont les suivants : 1, 2, 3, 4, 5, 6, 7, 8, 9 (tableau 2).
Étude de la robustesse de la méthode
La norme NF EN ISO 15189:2012 [3] (au § 5.6.2.1) précise qu’il faut vérifier la qualité prévue des résultats. Dans un premier temps, il est nécessaire de déterminer la robustesse de la méthode utilisée (tableau 3).
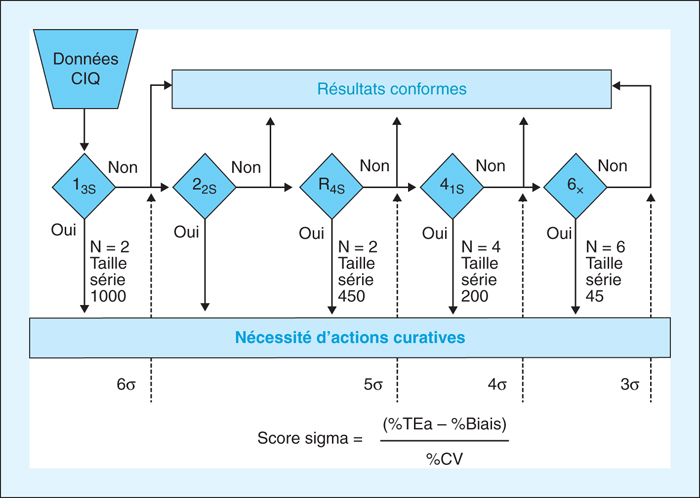
L’approche 6 sigma (sigma = (Et-Biais)/CV), qui n’est pas un objectif en soi, est un outil qui permet d’évaluer la robustesse de la méthode [5]. Le niveau de sigma est calculé à partir de l’erreur totale admissible (Eta) que choisit le laboratoire, des CV et des biais qui sont des données objectives caractéristiques de chaque méthode. La difficulté réside dans le choix de l’Eta qui peut conditionner grandement le résultat du niveau de sigma [6]. Actuellement un débat existe sur la formule de calcul du sigma avec la prise en compte ou non du biais [7].
D’autres moyens peuvent être utilisés pour évaluer la robustesse d’une méthode : suivi du rapport répétabilité/reproductibilité [4], fréquence des ré-étalonnages nécessaires, fréquence de reciblage des CIQ, etc.
Le LBM peut aussi exploiter les résultats du groupe de pairs (justesse), les résultats de sa technique au travers des comptes rendus des OCIL et éventuellement les résultats des confrontations avec d’autres laboratoires en dehors du cadre des OCIL.
Le résultat du calcul du niveau de sigma est surtout utilisé pour définir la stratégie de passage des CIQ : voir infra (§2.5), site internet Westgard [8]. L’approche 6 sigma est adaptée pour les grandes séries.
Stratégie de passage des CIQ : définition de la série et définition des évènements critiques (tableau 4)
Le laboratoire doit mettre en évidence ce qui est susceptible de modifier la stabilité de la méthode. Le laboratoire identifie les évènements critiques ou points de contrôles critiques (critical control point quality control) : l’étalonnage, une maintenance impactante (remplacement de pièces, réglages, etc.), certains types de panne, un changement de lot de réactifs ou d’étalons, etc. Le laboratoire peut ainsi définir les évènements qui vont clôturer la série [9].
La stratégie de passage des CIQ est basée sur :
- –la définition de leur fréquence de passage : risques 2 et 5 ;
- –les niveaux utilisés : risques 1 et 2 ;
- –le positionnement dans la série (étalonnage, nombre de dosages) : risques 2 et 7 ;
- –les évènements potentiellement impactants qui surviennent : risques 2, 3, 4, 6 ;
- –la définition des limites acceptables et le choix des règles d’interprétation des cartes de contrôles : risques 1, 2, 8.
Pour la définition de la série, il faudra prendre en compte également :
- –la stabilité de l’échantillon (analyse de risques à réaliser lorsque la stabilité de l’analyte ne permet pas toujours un redosage de l’échantillon à distance, bicarbonates par exemple) ;
- –la criticité du paramètre si résultat rendu dans le cadre de l’urgence (troponine, D-dimères, NFP, etc.) avant passage d’un CIQ pour clôturer la série ;
- –la robustesse de la technique utilisée ;
- –les éventuelles recommandations du fournisseur.
Le laboratoire encadre donc la série avec des CIQ ou d’autres moyens (voir §4.3 ci-dessous). Néanmoins, « en fin de journée », après le passage d’un CIQ de fin de série, il est acceptable de considérer que la méthode est sous contrôle et stable pendant un temps relativement court qui reste à définir par le LBM (quelques heures selon le retour d’expérience des auteurs), pour un nombre limité de dosages (moins de 50, selon le retour d’expérience des auteurs) en tenant compte de la robustesse de la méthode.
Place des maintenances préventives dans la stratégie de passage des CIQ
Voir risque n̊ 4
Certaines maintenances préventives peuvent avoir un impact sur la méthode (à documenter avec le fournisseur) : cette information est importante à prendre en compte afin de maîtriser son équipement.
Le moyen le plus simple pour maîtriser l’impact de ces maintenances sur la stabilité du système analytique est de passer des CIQ avant et après ces maintenances mais le laboratoire peut aussi utiliser d’autres moyens (redosages d’échantillons patients, etc.) (tableau 5).
Place de la maintenance curative
Voir risque n̊ 6
La maintenance curative est potentiellement impactante et dans ce cas, elle « clôture » une série de manière inopinée : le laboratoire doit vérifier l’absence de dérive de la méthode avant la panne (étude d’impact) (tableau 6).
Nombre de tests dans la série et fréquence des CIQ
Voir risque n̊ 8
Le laboratoire devra déterminer la fréquence de passage des CIQ et la taille de la série (nombre de dosages pour un paramètre entre 2 résultats de CIQ) (tableau 7).
Le calcul du niveau de sigma est un des moyens (voir §2.5 ci-dessous) d’évaluation de la robustesse de la méthode mais d’autres éléments sont à prendre en compte dans une analyse de risques :
- –la criticité clinique du paramètre ;
- –le délai de diffusion et d’exploitation des résultats ;
- –la possibilité de ré-analyse des échantillons (délais pré-analytiques respectés), lorsque cela est réalisable (impossible pour certaines analyses comme les gaz du sang par exemple).
Des auteurs, dans des publications récentes, proposent d’adapter la taille de la série et le choix des règles de Westgard en fonction du niveau de sigma [10, 11].
Le choix des CIQ et des limites acceptables
Exigences normatives
Voir risques n̊ 1 et 2
Les contrôles internes de qualité (CIQ) doivent répondre aux exigences suivantes de la norme NF EN ISO 15189:2012 (§5.6.2.2) :
- –matériaux se comportant par rapport au système analytique le plus fidèlement possible comme des échantillons patients. La non-commutabilité des CIQ n’est pas rédhibitoire s’ils sont plus sensibles aux problèmes analytiques que les échantillons de patients (pour le suivi de la reproductibilité intra-laboratoire) ;
- –revue régulière des résultats en fonction de la stabilité de la méthode et du risque d’impact sur le patient en cas de résultat erroné ;
- –niveaux de concentrations des matériaux de contrôle proches des seuils de décision médicale.
CIQ indépendant du fournisseur ?
La norme NF EN ISO 15189:2012, §5.6.2.2 - note 2 (non opposable) (2), recommande des CIQ indépendants du fournisseur pour maîtriser un risque de non-détection d’une dérive de la méthode par un défaut de commutabilité ou de fausse alarme en cas de changement de réactif. Néanmoins, en cas d’intervention du fournisseur, les CIQ fournisseur sont nécessaires pour une analyse critique pertinente (tableau 8).
Nombres et niveaux des contrôles
Le nombre et les niveaux des CIQ sont également à définir : le CIQ doit explorer toute l’étendue de mesure mais aussi les seuils décisionnels. Il doit également permettre de vérifier l’étalonnage (tableau 9).
Limites acceptables
Voir risque n̊ 8
L’objectif principal est de détecter des anomalies et des tendances (décalage et dérive) et de vérifier que la qualité prévue des résultats est atteinte.
Les limites acceptables (le terme de « spécification de performance analytique » est employé dans les dernières recommandations européennes) doivent être les plus proches des performances réelles de la méthode afin de détecter des tendances, des décalages ou des dérives. Attention à ne pas confondre dérive et décalage (drift et shift). Une dérive est une augmentation (ou une diminution) régulière des résultats. Un décalage correspond à un écart constant par rapport à la moyenne.
Le laboratoire choisira pour chaque analyse, un CV (cf. « qualité prévue » § 5.6.2.1. de la norme NF EN ISO 15189:2012) en fonction des performances des lots précédents. Ces CV sont les CV propres au laboratoire : CVLT (CV long terme) prenant en compte toutes les sources de variabilité de la méthode. Ils sont utilisés pour définir les limites acceptables des cartes de contrôle.
Dans un second temps, pour le suivi des performances, le laboratoire comparera les CV qu’il a obtenus à des CV de référence. Les référentiels des CV peuvent être :
- –CV issus des variations biologiques (par exemple : EuBIVAS) ;
- –CV issus des données fournisseur ;
- –CV issus des groupes de pairs (CIQ externalisés) ;
- –CV issus des recommandations de sociétés savantes françaises ou étrangères ;
- –CV issus des organismes de contrôle inter-laboratoires ou OCIL associatifs ou industriels (les principaux OCIL associatifs français sont regroupés en France au sein de la FAEEQ (voir §8 ci-dessous)).
En pratique et lorsque les règles de Westgard sont utilisées (tableau 10)
La perspective envisagée dans ce tableau permet au laboratoire de s’assurer que les performances analytiques restent stables et sous contrôle. Cette proposition ne prend pas en compte l’impact clinique potentiel (ou l’absence d’impact) en cas de performance analytique non satisfaisante (voir § 5, Étude d’impact post CIQ hors limites acceptables).
Une interprétation complémentaire éventuelle (en fin de série) peut être mise en œuvre afin de prendre en compte l’impact clinique potentiel. Deux niveaux de lecture sont alors mis en œuvre : des limites acceptables purement analytiques et des limites de tolérance en lien avec un risque médical.
Cartes de contrôle
Les objectifs des règles de Westgard sont :
- –de détecter une erreur analytique systématique ou aléatoire ;
- –de stopper la production des résultats en cas d’erreur avérée ;
- –d’estimer le biais (l’erreur) induit sur les résultats déjà rendus afin d’évaluer l’impact sur les résultats des patients et en termes statistiques d’obtenir une :
- •probabilité de détection de l’erreur – Ped (probability of error detection) supérieure à 90 % : elle mesure le pourcentage de chance de détecter une erreur en présence d’un dérèglement de la méthode analytique ; cette probabilité doit être la plus élevée possible ;
- •probabilité de faux rejet – Pfr (probability of false rejection) inférieure à 5 % : elle correspond au risque de rejeter une série en l’absence de tout dérèglement de la méthode analytique : cette probabilité doit être la plus faible possible.
Certaines recommandations internationales peuvent permettre de choisir a minima les règles de rejets applicables sans tenir compte de la robustesse de la méthode (risque de surqualité) (tableau 11) [13].
D’autres auteurs adaptent les règles de Westgard à la taille de la série en fonction du niveau de sigma [10] et des probabilités de détection des erreurs [14, 15] (figure 1).
EWMA
Afin d’améliorer les détections des faibles dérives, Roberts en 1959 [16] propose le procédé EWMA (exponentially weighted moving average) basé sur la statistique bayésienne [17]. Le principe de l’EWMA est simple : chaque nouvelle valeur de CIQ est pondérée par les valeurs précédentes. Il s’agit d’une méthode de lissage des nouveaux résultats obtenus par une moyenne glissante à pondération exponentielle, c’est-à-dire qui privilégie les points les plus récents aux dépens des anciens. En pratique, cela permet une détection plus précoce des faibles dérives [18]et une meilleure détection des erreurs systématiques [19].
Les changements de lots réactifs et de lots de CIQ
Voir risques n̊ 1 et 2
Exigences normatives NF EN ISO 15189:2012 (§5.3.2.3)
NF EN ISO 15189:2012 (§5.3.2.3) : Essai d’acceptation : « Chaque nouvelle formulation de trousse de réactifs prêts à l’emploi résultant de modifications de réactifs ou de procédure, un nouveau lot de fabrication ou une nouvelle expédition doit être vérifiée en matière de performance avant leur utilisation. »
Pour vérifier les performances des réactifs et des consommables, le laboratoire établit une stratégie d’acceptation à partir d’une analyse de risques, basée par exemple sur les données issues des fiches fournisseurs, les certificats de conformité et la stratégie de passage de contrôles de qualité (SH REF 2 [3]).
À chaque changement de lot d’échantillons de contrôle (CIQ), le laboratoire veille à anticiper l’établissement des valeurs cibles et des seuils d’interprétation. Ceux-ci sont déterminés selon des essais probatoires définis par le laboratoire en fonction de la spécificité de l’analyte et de la durée de validité du lot. Pendant cette période, la conformité de la technique est assurée par le lot de contrôle en cours.
La moyenne des résultats obtenus permet de déterminer la valeur cible initiale, les seuils d’interprétation des nouvelles cartes de contrôle seront réajustés si nécessaire.
Le nombre de déterminations préliminaires sera adapté à la période d’utilisation du lot de CIQ (période très courte de 1 à 2 jours pour l’hématologie par exemple à plus longue en cas de lot de CIQ utilisé pendant 1 an (certains CIQ utilisés en hémostase ou biochimie par exemple).
Une autre approche consiste à utiliser des tests statistiques associés basés sur le théorème de Bayes [17].
Essais d’acceptation
L’objectif des essais d’acceptation est de s’assurer que le produit (réactif, CIQ, consommable…) est conforme aux besoins du laboratoire avant d’autoriser son utilisation, suffisamment tôt pour pouvoir commander un nouveau lot et organiser une nouvelle expédition, (ou prévoir un back up ou une sous-traitance, etc.) dans le but d’éviter toute rupture de production qui peut entraîner une perte potentielle de chance pour les patients en cas de paramètres critiques.
La stratégie est à définir pour chaque analyte en fonction des données disponibles, de la durée de conservation du produit (CIQ en hématologie par exemple) et des risques identifiés.
Nouveau lot de CIQ
Les recommandations de la littérature (CLSI [9] et Tietz [12]) sont :
- –la détermination de la valeur cible par le laboratoire (10 mesures sur 10 jours) ;
- –la détermination de l’écart type par le laboratoire (20 mesures) ;
- –le calcul des limites à partir des moyenne et écart type du laboratoire (ou utilisation d’un écart type défini par le laboratoire issu de son expérience).
Les données seront à affiner après quelques semaines pour obtenir des valeurs prenant en compte une plus grande variabilité, comme les maintenances préventives, les étalonnages, etc. (tableau 13).
Nouveau lot de réactif
L’analyse de risques initiale est à réaliser, classiquement lors de la vérification/validation de la méthode [20] :définir l’impact du produit (diluant, réactif, étalon, etc.). Le produit est-il critique et quelle est sa robustesse ?
L’analyse d’échantillons « frais » provenant de patients est la méthode de référence mais d’autres moyens peuvent être utilisés (moyenne patients, pools, etc.) ; le passage isolé d’un CIQ pour valider un nouveau lot de réactif n’est pas recommandé [10].
Néanmoins pour les substrats (glycémie, cholestérol, etc.), la commutabilité des CIQ semble suffisante. En hormonologie, des effets lots sont fréquents et la confrontation externe du CIQ n’est pas toujours pertinente. Pour les marqueurs tumoraux, la commutabilité des CIQ étant fréquemment en défaut, le passage d’échantillons « frais » provenant de patients est vivement conseillé (tableau 14).
Nouvelle formulation de réactif, nouvelle référence ?
En cas de nouvelle formulation de réactif (et de nouvelle référence chez le fournisseur), le laboratoire doit réaliser une étude d’impact, à partir de l’étude de la documentation du fournisseur :
- –simple étude bibliographique qui conclut à l’absence d’impact ou impact documentaire simple : changement de conditionnement, changement des modalités de stockage sans impact sur la méthode, etc. ;
- –vérification uniquement des modifications : comparaison avec les résultats antérieurs en cas de nouveau raccordement de l’étalon. Étude de l’impact sur la justesse ou l’exactitude et éventuellement sur les valeurs de référence ;
- –vérification de méthode complète en cas de changements majeurs (dans le cadre de gestion de la portée flexible).
Les détections de tendance, les indicateurs qualité
Détection des tendances
Voir risque n̊ 8
La norme NF EN ISO 15189:2012 § 5.6.2.3 recommande que « Les données de contrôle qualité doivent être revues régulièrement pour détecter les tendances de réalisation d’analyses qui peuvent indiquer des problèmes dans le système d’analyse. Si de telles tendances sont observées, des actions préventives doivent être prises et enregistrées. ».
Rappel : Quelles sont les bonnes pratiques pour le calcul du CV du laboratoire ? Le principe du contrôle de qualité par l’utilisation des cartes de Levey-Jennings revient à déterminer si une valeur appartient à la population « habituelle » ou à une population différente (moyenne décalée et/ou CV plus élevé) apparue à la suite d’un dérèglement analytique.
L’estimation de paramètres statistiques (moyenne, écart type ou CV) consiste à estimer les paramètres de la population à partir d’un échantillon statistique issu de celle-ci.
Si l’échantillon statistique contient des valeurs issues de deux populations différentes, le calcul n’a plus de sens.
La dispersion « habituelle » d’une méthode correspond aux causes qualifiées de « communes », les valeurs de CIQ à conserver sont celles qui reflètent les performances de la méthode.
Si un dérèglement analytique est détecté par des valeurs hors contrôle (violation des règles 1-3S, 2-2S, R-4S), c’est qu’une cause « spéciale » est apparue et par conséquent, que les valeurs observées dans ces conditions n’appartiennent pas à la population « habituelle ». Les résultats de patients ne sont pas rendus tant que le dérèglement n’est pas corrigé.
Et c’est pourquoi il semble logique et cohérent de ne pas inclure ces valeurs dans le calcul d’un CV qui doit représenter le fonctionnement « habituel » du laboratoire.
En revanche, si le redosage des mêmes CIQ fournit des valeurs dans les limites acceptables, il est possible de conclure que la valeur précédemment hors contrôle fait partie de la population « habituelle ». Il s’agit du risque de première espèce (3 pour mille dans le cas de la règle 1-3S) et il est alors recommandé d’inclure cette valeur dans le calcul du CV. Les valeurs de CIQ à conserver pour calculer le CV de fidélité intermédiaire du laboratoire sont celles qui reflètent les performances réelles de la méthode et qui correspondent au traitement des résultats patients. Si le biologiste choisit de rendre les résultats des patients, par exemple au regard de l’erreur totale acceptable, il conservera la valeur du CIQ et inversement, il exclura les valeurs de CIQ en cas de rejet de la série associée. Cette attitude présente une double cohérence : statistique et vis-à-vis des objectifs analytiques.
Les erreurs grossières (inversion de niveaux de CIQ, fin de flacon, etc.) qui ne représentent pas la dispersion réelle de la méthode ne sont donc pas à prendre en compte.
Si la méthode est modifiée (changement de flacon de CIQ, re-étalonnage, etc.), il est logique d’exclure les points de CIQ non conformes du calcul du CV (données d’avant l’étalonnage ou du changement de flacon).
En revanche, si rien n’est modifié dans la méthode (redosage du même CIQ), il n’y a pas de justification à exclure ces points.
La principale des tendances à suivre est l’augmentation de la dispersion de la méthode qui est vérifiée grâce au CV. Le suivi des CV et leur conformité par rapport aux spécifications du laboratoire sont recommandés. Ce suivi est à réaliser régulièrement (en fonction de la robustesse de la méthode et de la fréquence de réalisation de la technique), et au moins tous les trimestres. En cas d’utilisation de plusieurs analyseurs pour la réalisation des mêmes analyses, la comparaison des CV des différents systèmes analytiques est recommandée.
Les analyses de tendance ont pour objectif de dépister au plus tôt toute dérive du système analytique et de mettre en œuvre des actions préventives si besoin (suivi de l’erreur aléatoire et systématique) :
- –une augmentation de la dispersion de la méthode (dérive de l’erreur aléatoire) pourra être objectivée par un suivi régulier de la méthode (fréquence à adapter pour chaque examen en fonction de la robustesse, de l’importance clinique, de la fréquence de réalisation, etc.). Pour les examens courants, un suivi régulier du CV est recommandé avec confrontation aux spécifications du laboratoire, selon une fréquence appropriée, le plus souvent mensuelle selon les examens afin de pouvoir agir rapidement si besoin ;
- –une augmentation de l’erreur systématique (ou biais de justesse) pourra être évaluée par la confrontation externe des résultats de CIQ (suivi régulier du Z-score ou IET (indice d’écart type) (tableau 15).
Reciblage des valeurs cibles des CIQ
Chaque laboratoire détermine la valeur cible qui correspond à la moyenne des valeurs obtenues pendant la période probatoire. Cette valeur est retenue comme valeur moyenne pour l’établissement de la carte de contrôle. Au cours de l’utilisation du lot d’échantillons de contrôle, la valeur cible peut être réajustée si nécessaire. La valeur cible retenue est calculée sur une période suffisamment étendue pour être significative [3].
En cas de décalage de plus d’un écart type (unité de mesure des cartes de contrôle), le laboratoire doit recibler la moyenne pour éviter le risque de faux rejet (perte de temps et retard de rendu) mais surtout de fausse acceptation (impact potentiel sur les résultats).
Tout « reciblage » est argumenté et précédé d’une étude sur les sources potentielles de variations (étalonnage, changement de lot de réactifs, maintenance, etc.). De plus le laboratoire devra s’assurer de l’absence d’erreur de justesse (avec la moyenne du groupe de pairs) ou d’exactitude (avec des EEQ) (tableau 16).
Moyens complémentaires de suivi des performances
Ces moyens peuvent compléter une stratégie de suivi de méthode en plus du CIQ, des confrontations externes des CIQ et des EEQ.
Ils ne sont pas obligatoires mais peuvent apporter un complément d’informations à condition d’être correctement exploités.
Moyenne patient
Le suivi de la moyenne des patients (appelée X-barM en hématologie) peut être un moyen complémentaire pertinent afin de détecter précocement une dérive ou un décalage d’un système analytique [22, 23]. Ce moyen présente l’avantage d’évaluer le décalage de la méthode à partir d’une matrice humaine et permet de pallier un défaut de commutabilité ou une détérioration du CIQ. Le suivi de la moyenne patient n’est pas pertinent pour tous les paramètres (marqueurs tumoraux, etc.).
En cas d’utilisation de la moyenne patient, une attention particulière devra être portée par le laboratoire :
- –à la taille de la population (qui permettra de calculer la moyenne et définira la valeur à reporter sur la carte de contrôle) et à l’exclusion de certains services critiques (dialyse, réanimation) : indicateur applicable seulement pour une population stable ;
- –aux limites acceptables définies par le laboratoire qui doivent être, a minima, comparables à celles des CIQ (même si statistiquement il ne s’agit pas des mêmes CV) ;
- –à la conduite à tenir en cas d’alarme (tableau 17).
Repasse d’échantillons de patients
L’analyse d’échantillons de patients plusieurs fois au cours de la journée (dans la limite de la stabilité des analytes) peut être utilisée par le laboratoire comme moyen de suivi des performances (« patient trace » ou « QC patient » ou encore « fil rouge ») avec un critère d’acceptabilité de 2,8 fois l’écart type de la méthode (Norme NF EN ISO 5725-6 (24)).
Ce moyen permet de pallier la non commutabilité de certains CIQ et peut aussi permettre de l’explorer (tableau 18).
Pool d’échantillons de patients
La constitution d’un pool d’échantillons provenant de patients peut être une solution retenue par le laboratoire. Ce choix peut être judicieux notamment en l’absence de CIQ et/ou en complément des CIQ pour explorer la commutabilité (marqueurs tumoraux, hormonologie, etc.). La stabilité de ce pool d’échantillons, souvent congelé, doit être explorée et documentée (tableau 19).
Paramètres « sentinelles »
Sur un analyseur multiparamétrique, certains paramètres sont plus sensibles que d’autres aux différents composants de l’appareil (pipetage de faible volume, longueur d’onde spécifique, temps réactionnel, etc.).
Un passage adapté de CIQ ou d’échantillons provenant de patients pour ces paramètres permet d’explorer la maîtrise de toutes les autres méthodes (d’autant que celles-ci sont robustes). La maîtrise du risque lié aux maintenances internes ou de celles du fournisseur (SAV) peut s’appuyer sur un panel de « paramètres sentinelles » afin de vérifier rapidement l’absence d’impact de la maintenance sur les différents composants de l’analyseur. La stratégie doit être argumentée, notamment sur la base de données fournies par le fabricant et sur l’évaluation de la robustesse des paramètres.
En cas de maintenance « lourde » imposant des étalonnages, cette stratégie ne peut pas être retenue (tableau 20).
Étude d’impact post-CIQ fin de série hors limites acceptables
Exigences normatives NF EN ISO 15189:2012 (§ 5.6.2.3)
En cas de non-respect des règles de contrôle qualité et si les résultats d’analyses sont susceptibles de contenir des erreurs cliniques significatives [12], les résultats doivent être rejetés et les échantillons des patients concernés doivent être de nouveau analysés après avoir corrigé l’erreur et vérifié la conformité de la performance avec les spécifications. Le laboratoire doit également évaluer les résultats des échantillons de patients qui ont été analysés après le dernier CIQ conforme.
Le SH REF 02 [3] précise que des seuils d’alarme et d’action sont à définir. Et qu’en cas de CIQ non conforme, le laboratoire s’attache à évaluer l’impact sur les résultats rendus depuis le précédent CIQ conforme.
Les dispositions
Le laboratoire doit avoir prévu l’étude d’impact potentiel d’une anomalie sur les résultats des patients dans ses dispositions (tableau 21).
Cause de la dérive de la méthode
Le laboratoire doit vérifier que le résultat de CIQ non conforme est le reflet d’un dysfonctionnement du système analytique et tout d’abord exclure la responsabilité du CIQ en vérifiant qu’un contrôle fraichement préparé est non-conforme ou qu’une ré-analyse d’échantillons frais provenant de patients et passés juste après un CIQ conforme conclut à des résultats non concordants (sinon risque de faux rejets avec des conséquences dommageables pour le laboratoire (en temps et en coût)).
Ensuite le laboratoire doit rechercher les causes du problème et déterminer l’étendue :
- –quels analytes concernés et à quels niveaux de valeurs ?
- –analyse des CIQ (tous les niveaux) ou ré-analyses d’échantillons provenant de patients ;
- –quelle est la cause probable ? : alarme analyseur explicite ou non ;
- –depuis quand le problème est-il survenu ? : revue des CIQ, alarmes analyseurs, moyenne mobile à la recherche d’une dérive.
Cette étude doit permettre de circonscrire l’étendue du problème et définir la liste des résultats de patients pouvant être impactés.
La stratégie de l’étude d’impact
Premier temps : Le choix des échantillons à ré-analyser dépend du nombre d’échantillons potentiellement impactés. Les résultats des échantillons ré-analysés sont exploités afin de vérifier que les limites de concordance analytiques sont inférieures à 2,8 CV (ISO 5725-6:1994) [24].
Second temps : En absence de concordance analytique, définition des limites de concordance cliniques :
- –les objectifs analytiques du consensus de Milan [25] s’appliquent (mais données issues d’études cliniques rares). En pratique l’erreur totale Ricos souhaitable [26] peut être utilisée ;
- –on peut aussi utiliser les limites acceptables des EEQ (qui définissent un impact clinique) et qui sont habituellement pertinentes.
En cas de dépassement des limites de concordance clinique, un rappel du compte rendu d’examens en cas de diffusion (amendement aux rapports d’essai selon le § 5.9.3 de la norme NF EN ISO15189:2012) est nécessaire. Cependant le SH REF 02 précise que les comptes rendus d’examens erronés sont remplacés, lorsque cela se justifie et en privilégiant le dialogue avec le clinicien (tableau 22).
La comparabilité multi-analyseurs
Voir risque n̊ 8
En cas d’utilisation de plusieurs systèmes analytiques pour la réalisation des mêmes analyses dans le laboratoire, la comparabilité des résultats fournis par les différents systèmes doit être assurée. Les EBMD sont également concernés par cette étude de comparabilité (entre différents EBMD et par rapport aux automates du laboratoire).
Fréquence de la surveillance
Le laboratoire doit définir une fréquence de surveillance : il n’y a pas de recommandation opposable pour cette fréquence mais le laboratoire devra argumenter à partir de son analyse de risques : il devra prendre en compte le nombre de dosages, la robustesse de la méthode, les conséquences d’une dérive d’un des systèmes, les autres moyens mis en place (CIQ aux limites acceptables communes, moyennes mobiles comparées, etc.) (tableau 23).
Matériel utilisable
Plusieurs possibilités s’offrent au laboratoire pour assurer cette comparabilité analytique : CIQ, échantillons frais issus de patients, pool d’échantillons de patients conservés, échantillons d’EEQ, études statistiques des résultats (moyenne des résultats des patients par exemple), etc. (tableau 24).
Indicateurs
Afin de détecter précocement une dérive d’un système, le laboratoire peut aussi utiliser plusieurs indicateurs : pourcentage de CIQ rejetés par système, suivi des CV de chaque système avec un rapport de CV < 2 (proposition des auteurs) entre les différents systèmes, pourcentage de rejets des moyennes des patients normaux, pourcentage de rejets du « patient trace » (si utilisé), nombre d’EEQ non conformes, pourcentage de rejets des ré-analyses des échantillons des patients (critères de validation de repasse non respectés), pourcentage d’alarmes qualitatives en hématologie par analyseur.
Ces indicateurs constituent des alertes précoces pouvant entraîner une enquête.
Évaluation de l’impact clinique
En cas de différence analytique, le laboratoire devra définir l’impact clinique.
La décision finale de rappel de comptes rendus dépend de la différence critique entre 2 résultats, c’est-à-dire la définition de l’impact clinique.
Les différentes formules et concepts que le laboratoire peut utiliser sont résumés dans le tableau 25.
Remarque : Les limites acceptables définies par les OCIL sont définies selon le consensus de Milan [25] et dépendent de l’analyte concerné : elles peuvent être basées sur l’expérience de l’OCIL, sur l’erreur totale Fraser, etc. (tableau 26).
La confrontation externe des CIQ
La confrontation externe des CIQ est un outil complémentaire [27] : qui permet de :
- –assurer le suivi de la justesse de la méthode analytique par rapport à la moyenne des pairs (justesse) et de la fidélité intermédiaire par rapport à l’écart-type ou au CV des pairs (à condition qu’il soit calculé comme un écart-type ou CV intralaboratoire) ;
- –assurer un reciblage en cas de décalage interne (ou de paramétrer une carte de contrôle en cas d’absence de « période probatoire ») ;
- –obtenir les caractéristiques de la méthode (CV, CV long terme, Biais, IM, etc.) (tableau 27).
Les EEQ
Choix d’une EEQ
Les critères de choixd’un EEQ sont présentés dans le tableau 28 (pratique recommandée). La commutabilité [29-31] est souhaitable mais l’information sur cette donnée est parcellaire. Le laboratoire pourra analyser les rapports des OCIL afin d’estimer cette commutabilité (en comparant les écarts entre les différentes méthodes selon la nature des échantillons de contrôle par exemple).
Passage des EEQ
Les laboratoires soumettent à une évaluation externe de la qualité chaque système analytique qu’ils utilisent [32].
Le laboratoire doit analyser les matériaux de contrôle des EEQ comme un patient (une seule fois) et ne doit pas répéter la mesure sauf si cela est prévu dans ses dispositions (règles de redosage automatique).
En cas de dosage des EEQ sur plusieurs analyseurs, il convient de rendre le résultat d’EEQ obtenu pour chaque analyseur sans correction du résultat.
Limites acceptables et interprétation des résultats
Trois types de valeurs cibles sont exploitables pour les évaluations : moyenne des valeurs toutes méthodes confondues, par groupes de pairs et valeur obtenue avec la méthode de référence (si existe et est accessible).
Pour chaque évaluation, les limites acceptables sont définies par les OCIL (besoins cliniques, état de l’art [33] ou variations biologiques).
Chaque OCIL définit ses propres limites acceptables en s’appuyant sur les données de la littérature (tableau 29).
En cas d’absence d’EEQ disponible
Dans les rares cas d’absence d’EEQ disponibles (situation rare pour les analyses de pratique courante), le laboratoire doit démontrer l’exactitude des résultats fournis (tableau 30).
L’interprétation des IM et leur calcul
Attention à ne pas confondre erreur totale et incertitude : l’erreur totale est la différence entre la valeur mesurée et la valeur vraie et l’incertitude est la quantification du doute autour du résultat mesuré [34].
L’incertitude de mesure est particulièrement importante en cas d’interprétation du résultat en fonction d’un seuil décisionnel entraînant une conséquence pour la conduite médicale vis-à-vis du patient (taux d’hémoglobine et transfusion, dosages de médicaments et adaptation de la posologie, CDT et commission du permis de conduire, etc.).
En pratique, classiquement, le calcul de l’incertitude repose sur la combinaison quadratique de 2 termes : la dispersion et le biais. Pour le biais, le biais moyen (avec l’écart-type du biais) ou le biais max peuvent être utilisés (SH GTA 14) [34].
La comparaison de l’incertitude de mesure à des spécifications est une obligation normative (selon le § 5.5.1.4 de la norme NF EN ISO15189:2012 [2]). Si les 2 composantes de l’incertitude de mesure (dispersion et biais), suivies régulièrement n’ont pas varié, le suivi peut être espacé. Néanmoins, les OCIL qui fournissent une estimation de l’incertitude de mesure font un bilan annuel avec surtout une comparaison de tous les laboratoires participants. Enfin, le choix des exigences de performances est difficile : l’erreur totale n’est pas rigoureusement statistiquement comparable et il existe peu d’autres données récentes dans la littérature.
Le suivi des incertitudes de mesure reste un outil interne basé sur le suivi des performances analytiques des paramètres.
En pratique [35, 36] : tableau 31
Tests unitaires simplifiés (TUS), examens de biologie médicale délocalisés (EBMD) et contrôles de qualité
L’analyse de risques doit être réalisée spécifiquement pour ces tests et doit prendre en compte notamment le volume de dosages réalisés et l’exploitation des résultats.
Si le CIQ est utilisable sur le TUS ou pour l’EBMD, les recommandations sont comparables(tableau 32).
Conclusion
Ce document a fait l’objet d’une version publique et les auteurs ont reçu près d’une centaine de propositions d’amélioration issues de tout type de laboratoires (laboratoire de biologie médicale de CHU, de CHG, de laboratoires de référence et laboratoires de biologie médicale privés).
Ce document sera revu régulièrement afin de s’adapter aux évolutions scientifiques et techniques.
Ce document tente de définir la stratégie de contrôle de qualité (interne et externe), basée sur une analyse de risques et constituant la validation/vérification continue de la méthode.
Cette stratégie, utilisant des méthodes statistiques doit permettre de produire des résultats validés afin de répondre aux besoins des prescripteurs.
Liens d’intérêts
S. Albarede : coordonnateur des programmes EEQ du CTCB (salarié). J.-L. Galinier : dirigeant bénévole des associations CTCB et FAEEQ. M. Kuentz : intervention ponctuelle auprès de Biomérieux. J.-P. Siest : président de Biologie Prospective. Les autres auteurs déclarent ne pas avoir de lien d’intérêts en rapport avec cet article.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International