Annales de Biologie Clinique
MENUÀ propos d’un cas de leucémie plasmocytaire primitive à IgE Volume 77, numéro 5, Septembre-Octobre 2019

Le myélome multiple est une prolifération médullaire maligne de plasmocytes responsables de la sécrétion d’une immunoglobuline (Ig) monoclonale complète ou d’une chaîne légère libre kappa ou lambda. Le myélome à IgE est extrêmement rare (moins de 0,01 % des cas de myélome) [1]. Les manifestations cliniques ont peu de spécificités par rapport aux types plus usuels de myélome.
La leucémie à plasmocytes est une forme rare et grave du myélome multiple [2]. Elle représente 2 à 4 % des myélomes selon la classification OMS 2017 [3].Elle est définie par une plasmocytose sanguine supérieure à 2 G/L ou 20 % des leucocytes [4].Il en existe deux variantes : la leucémie à plasmocytes primitive ou LPp (60 % des cas), survenant de novo chez des patients sans signe préalable de myélome et la leucémie à plasmocytes secondaire ou LPs (40 % des cas), évolution leucémique d’un myélome multiple connu en rechute et/ou réfractaire au traitement.
Nous rapportons ici un cas de leucémie plasmocytaire de novo à IgE.
L’observation
Il s’agit d’un patient âgé de 66 ans, admis pour découverte d’une insuffisance rénale et d’une hypercalcémie, associées à des douleurs osseuses lombaires et à une altération de l’état général. Le patient est suivi pour un diabète de type 2 et ne présente pas d’autre antécédent. L’examen clinique met en évidence une pâleur cutanéomuqueuse isolée sans autres signes associés notamment pas d’adénopathie, ni d’hépato-splénomégalie.
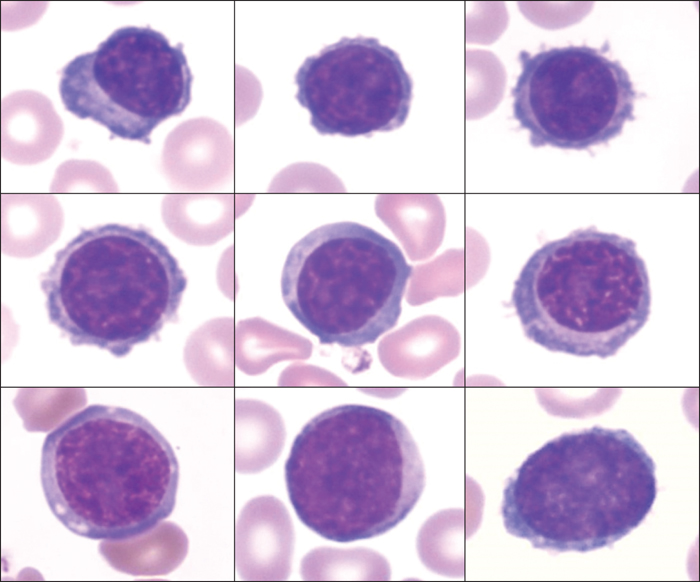
L’hémogramme montre une anémie à 8 g/dL, normocytaire normochrome arégénérative, des plaquettes à 190 G/L et une hyperleucocytose à 14,1 G/L. La lecture du frottis sanguin montre la présence de 22 % de plasmocytes nettement dysmorphiques : petite taille, rapport nucléocytoplasmique élevé, noyau souvent en position centrale, chromatine parfois intermédiaire, et nucléolés (figure 1).
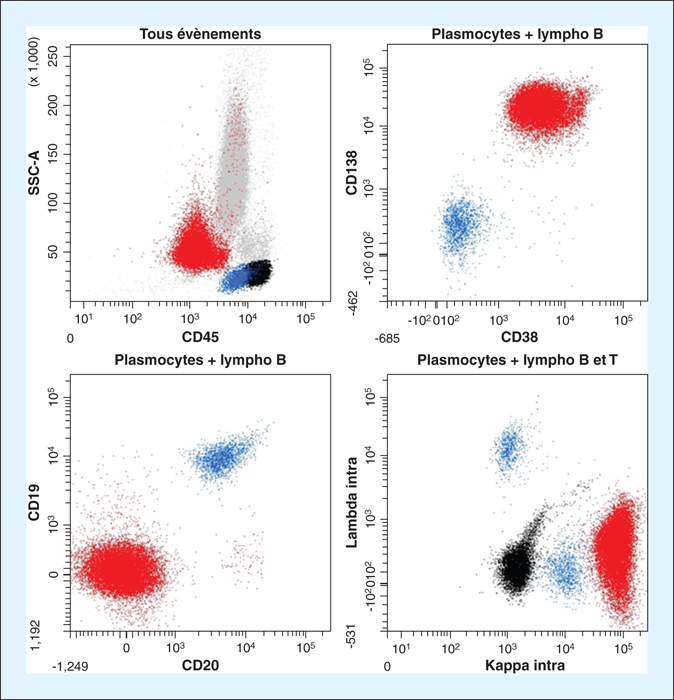
L’immunophénotypage sanguin confirme la présence d’un contingent de plasmocytes pathologiques évalué à 27 % des leucocytes. Ces cellules sont CD138+ CD38+ d’intensité diminuée, les CD45 et CD19 sont négatifs, le CD20 n’est pas exprimé, enfin on retrouve une monotypie kappa sur les marquages intracellulaires. Le diagnostic évoqué est celui de leucémie à plasmocytes (figure 2).
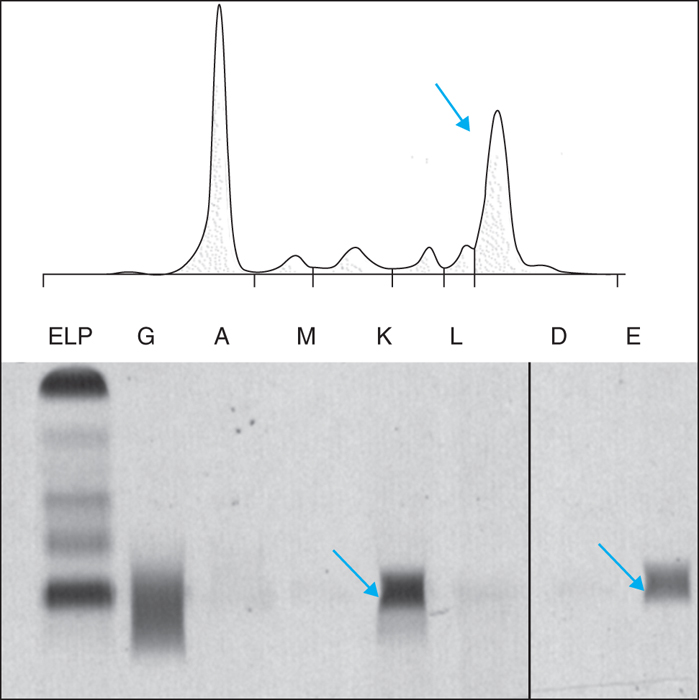
La protidémie est à la limite supérieure de la normale (80 g/L). L’électrophorèse des protéines sériques sur gel d’agarose montre la présence d’un pic monoclonal migrant au niveau des gammaglobulines quantifié à 25 g/L (figure 3). En présence de ce pic, un immunotypage est réalisé en utilisant des anti-sérums anti-immunoglobulines IgG, IgA, IgM et anti-chaînes légères lambda et kappa. Celui-ci révèle une soustraction du pic monoclonal uniquement en présence d’antisérum anti-chaînes légères kappa. L’immunofixation des protéines sériques sur gel d’agarose avec des anti-sérums anti-immunoglobulines IgG, IgA, IgM et chaînes légères lambda et kappa, retrouve une bande étroite unique au niveau des chaînes légères kappa. Une deuxième immunofixation utilisant des antisérums anti IgD et IgE est alors réalisée, permettant d’objectiver une gammapathie monoclonale à IgE (figure 3).
Il existe une répression de synthèse des autres classes d’immunoglobulines (dosages pondéraux des IgA, IgG, IgM respectivement à 0,23, 3,3 et 0,06 g/L). Le dosage sérique des chaînes légères libres (CLL) kappa est élevé à 1 302 mg/L avec un ratio kappa/lambda à 1 302. L’analyse biochimique des urines révèle une protéinurie à 0,21 g/L, composée d’un tiers de chaînes légères monoclonales kappa (protéinurie de Bence Jones).
Le bilan biochimique objective une insuffisance rénale aiguë (créatinine à 279 μmol/L avec une valeur basale à 70 μmol/L) et une hypercalcémie maligne à 4,31 mmol/L (calcémie corrigée à 4,48) avec un retentissement neurologique (confusion, ralentissement psychomoteur) et cardiaque (raccourcissement du QTc à 350 ms et bloc auriculo-ventriculaire type 1) nécessitant la prise en charge du patient en soins intensifs.
L’exploration radiologique par IRM du rachis entier retrouve une infiltration myélomateuse diffuse de l’ensemble du rachis avec fracture du corps vertébral L3. L’étude du liquide céphalorachidien ne montre pas d’infiltration myélomateuse. On note une élévation de la β2-microglobuline à 10 mg/L, une hypoalbuminémie à 33 g/L, et des LDH normales (431 U/L, valeurs normales : 240-480 U/L).
Le myélogramme, largement hémodilué, montre une plasmocytose à 33 % faite de plasmocytes dystrophiques d’aspect semblable aux plasmocytes circulants. L’analyse cytogénétique des plasmocytes n’est pas disponible.
Au total, le diagnostic retenu est celui d’une leucémie à plasmocytes à IgE kappa de novo.
Le patient est classé selon le score ISS (International staging system) en stade III (β2-microglobuline > 5,5 mg/L). Un traitement par bortézomib-cyclophosphamide-dexaméthasone est démarré au premier cycle compte tenu de l’insuffisance rénale, poursuivi par bortézomib-lénalinomide-dexaméthasone, et suivi d’une intensification par melphalan et d’une autogreffe.
Le point de vue du clinicien
Chez notre patient, la LPp de novo est découverte à un stade avancé avec une insuffisance rénale aiguë et une hypercalcémie maligne nécessitant son hospitalisation en réanimation. L’exploration biologique retrouve alors une gammapathie à IgE.
Le myélome à IgE est une maladie extrêmement rare, décrite pour la première fois en 1967 et représentant moins de 0,01 % des cas de myélome multiple [1].Ilsurvient à un âge médian de 55-60 ans, avec une nette prépondérance masculine. Les manifestations cliniques ont peu de spécificités par rapport aux types plus usuels de myélome, en dehors de la constatation assez fréquente, mais non constante, de formes leucémiques primitives ou secondaires. Le pronostic est très péjoratif avec une survie moyenne de 33 mois [5].
La leucémie à plasmocytes primitive ou de novo est également une maladie très rare. Son incidence en Europe est d’environ 0,04 cas pour 100 000 habitants par an, survenant à un âge plus jeune que le myélome avec une moyenne d’âge de 52-65 ans [2, 4, 6].Les deux entités de leucémie à plasmocytes présentent un tableau clinique agressif avec de fréquentes localisations extra-médullaires (jusque 20 % des cas) [6], au premier rang desquelles des atteintes hépatiques et spléniques (52 et 40 % des cas), les adénopathies étant plus rares. Un envahissement viscéral peut être observé au diagnostic (notamment pleuro-pulmonaire, neuro-méningé, testiculaire). Ici, le patient ne présentait pas de syndrome tumoral ni d’atteinte viscérale.Les lésions ostéolytiques sont moins fréquentes que dans le myélome multiple (dans 81 % des myélomes, contre 35 % des LPp et 53 % des LPs) [7].Le pronostic de la LPp est très péjoratif avec une médiane de survie globale inférieure à 12 mois [4].
Dans le myélome, l’évaluation du pronostic est basée sur le calcul du score ISS (2005) permettant une stratification du risque à partir de deux paramètres au diagnostic : le taux de la β2-microglobuline (β2m) reflétant la masse tumorale et le taux sérique d’albumine (tableau 1).Le score ISS révisé (2018) intègre quant à lui, la valeur des LDH, et 2 anomalies cytogénétiques de mauvais pronostic, la del(17p) et la t(4;14) (tableau 2) [8].
Comme pour le myélome, le traitement est basé sur la polychimiothérapie comportant des inhibiteurs de protéasome (bortézomib, carfilzomib) et/ou des agents immunomodulateurs (lénalidomide, thalidomide, pomalidomide), suivie d’une ou plusieurs autogreffes [9]. Le traitement du myélome a bénéficié récemment de nombreuses avancées thérapeutiques, avec l’arrivée de nouvelles molécules ciblées, telles que le daratumumab, premier anticorps monoclonal anti-CD38 ayant obtenu une autorisation de mise sur le marché pour le traitement des patients atteints de myélome en rechute. Cependant il existe peu de données sur l’efficacité de ces nouveaux médicaments dans la LPp, la littérature étant souvent basée sur des rapports de cas ou de petites séries rétrospectives [10].
Le point de vue du biologiste
Les gammapathies à IgE sont exceptionnelles et la recherche spécifique des IgE ne fait habituellement pas partie de l’immunofixation (IF) ou immunotypage (IT) de première intention, lors de l’exploration d’un pic monoclonal à l’électrophorèse des protéines. L’isotype IgE doit être évoqué devant tout pic monoclonal sur l’électrophorèse, avec un marquage isolé par les anti-sérums anti-chaînes légères kappa ou lambda, sans marquage avec les anti-sérums IgG, IgM et IgA, à l’IF et/ou IT. Dans ce cas de figure, il est nécessaire de réaliser en complément un marquage par des anti-sérums anti IgD et IgE.
Dans la LP, les cytopénies (anémie et thrombopénie) ainsi que l’insuffisance rénale et l’hypercalcémie sont souvent plus sévères que dans le myélome multiple [7]. Le diagnostic de la LP est biologique, il repose tout d’abord sur les données de l’hémogramme et le frottis sanguin coloré au May Grünwald Giemsa qui montre une plasmocytose sanguine supérieure à 2 G/L ou un taux de plasmocytes circulants supérieur à 20 % des leucocytes. Les plasmocytes ont parfois un aspect pseudo-lymphocytaire les rendant plus difficilement identifiables au frottis sanguin et le recours à l’immunophénotypage dans ces formes ambiguës est alors indispensable au diagnostic. L’immunophénotypage des plasmocytes de LP se distingue du myélome multiple par une expression plus fréquente du CD20 et moins fréquente du CD56 (négatif dans 80 % des LP). Le CD28, marqueur d’évolutivité tumorale dans le myélome, est plus fréquemment exprimé dans les LPs (92 % des LPs et 33 % des LPp) [4, 11]. Un certain nombre de molécules d’adhésion seraient impliquées dans le processus de leucémisation, parmi lesquelles le CD56 qui joue un rôle important dans l’ancrage des plasmocytes au stroma médullaire. L’absence d’expression du CD56 dans la LP pourrait favoriser la circulation des plasmocytes et leur migration vers des sites extramédullaires [11].
Le myélogramme ou la biopsie ostéomédullaire montre souvent une infiltration diffuse plasmocytaire variant de 50 à 100 % [4].Le caryotype des LP présenterait plus fréquemment des anomalies de haut risque (t(4 ;16), t(4 ;14) et del(17p)) par rapport aux myélomes multiples [12, 13].
Conclusion
La leucémie à plasmocytes et le myélome à IgE sont deux affections très rares et de pronostic très péjoratif. Dans les deux cas, le diagnostic biologique est au premier plan : l’identification d’une plasmocytose circulante > 20 % permet d’évoquer la leucémie à plasmocytes tandis que la mise en évidence d’une gammapathie IgE nécessite une vigilance particulière devant certains profils d’immunofixation ou immunotypage.
Liens d’intérêts
les auteurs déclarent ne pas avoir de lien d’intérêts en rapport avec cet article.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International