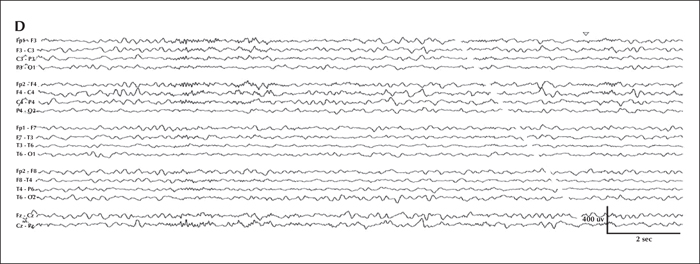
Serial EEG from initial presentation and the latest follow-up visit. Overnight video-EEG was performed during hospital admission for breakthrough seizures within two weeks of initial epilepsy diagnosis (2.5 months old) and captured an electroclinical tonic seizure characterized by unresponsiveness and staring for six seconds, followed by asymmetric (left>right) limb extension with vibratory stiffening. Electrographically, at onset (A), there was diffuse attenuation, subsequently maximal at the vertex, followed by rhythmic discharges that gradually spread to the bilateral central region; on the right more than the left (B). Sleep background at 2.5 months old (C) was significant for symmetric sleep spindles and intermittent epileptiform discharges maximal in the central vertex region. The most recent EEG was performed at 3 years of age (D) and background activity during sleep was significant for subtle relative left hemispheric attenuation, occasional multifocal epileptiform discharges, and asymmetric sleep spindles and vertex waves, higher in amplitude on the right.
MRI of the patient at different time points. (A) MRI performed at 2 months of age showing Rathke's cleft cyst (arrow) and asymmetric hemisphere size (left cerebrum smaller than right). (B) MRI performed at 11 months of age showing linear, radially-oriented T2 hyperintensities within the white matter with unclear significance (arrows). (C, D) MRI performed at 2 years of age shows right periventricular heterotopia (arrow in C) and abnormal perisylvian gyral configuration (C and arrow in D).
1 Division of Neurology, Cincinnati Child ren's Hospital Medical Center, Cincinnati, Ohio
2 Department of Pediatrics, Division of Genetics, Ann & Robert H. Lurie Children's Hospital of Chicago, and the Northwestern University Feinberg School of Medicine, Chicago, Illinois
3 Department of Pediatrics, Divisions of Neurology & Epilepsy, Ann & Robert H. Lurie Children's Hospital of Chicago, and the Northwestern University Feinberg School of Medicine, Chicago, Illinois, USA
* Correspondence: Charu Venkatesan
Division of Neurology,
Cincinnati Children's Hospital Medical Center,
3333 Burnett Avenue, MLC 2015,
Cincinnati, OH 45229, USA
Advances in genetic testing have led to the identification of increasing numbers of novel gene mutations that underlie infantile-onset epileptic encephalopathies. Recently, a mutagenesis screen identified a novel gene, SZT2, with no known protein function that has been linked to epileptogenesis in mice. Thus far, two clinical reports have identified children with different recessive mutations in SZT2 and varying clinical phenotypes. One case report described patients with epileptic encephalopathy and the other noted patients with cognitive deficiencies, but normal MRI and no epilepsy. This case report identifies novel mutations (a compound heterozygous frameshift and a nonsense variant) in the SZT2 gene with distinct clinical and radiographic findings relative to those previously reported. Our patient presented with intractable epilepsy at 2 months of age. Seizures were refractory to numerous antiepileptic medications and the patient finally achieved seizure cessation at age 3 years with a combination of divalproex and lamotrigine. Our patient had similar facial dysmorphisms (macrocephaly, high forehead, and down-slanted palpebral fissures) to a previous case with truncating mutation. While developmental delay and cognitive deficiencies were present, our case had unique MRI findings suggesting migrational abnormalities not previously reported in other cases.