Epileptic Disorders
MENUThe clinical pharmacology of traditional antiepileptic drugs Volume 16, numéro 4, December 2014


A number of drugs continue to be reasonably widely used some 50 years or more after they first became available for treating human epilepsy (Arzimanoglou et al., 2010). Details of their pharmacological profiles are provided below; the various substances are discussed largely according to when they were initially used in humans.
Phenobarbitone and its congeners
Phenobarbitone (phenobarbital), introduced to antiepileptic therapeutics in 1912, remains in extensive use worldwide, largely because of its favourable cost-effect ratio. Several phenobarbitone congeners have been developed as antiepileptic agents. Two, primidone (desoxyphenobarbitone) and N-methylphenobarbitone (mephobarbital), continue to be used clinically (Eadie and Hooper, 2002).
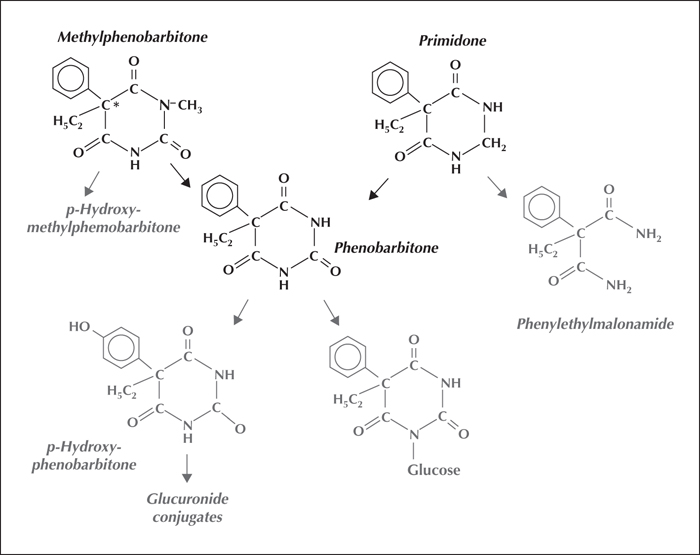
Phenobarbitone, in acid or its sodium salt form, is marketed for oral, intramuscular or intravenous use, and its congeners for oral use. Methylphenobarbitone and primidone are metabolised to phenobarbitone in humans (figure 1). Each possesses antiepileptic activity in its own right, but the resulting phenobarbitone probably provides the main antiepileptic effect during their continuing use in humans (Michelucci et al., 2009).
Mechanism of action
At a molecular level, phenobarbitone at therapeutically effective concentrations acts by prolonging the opening of post-synaptic cell membrane chloride ion channels within GABAA receptors, thus hyperpolarising the neuronal cell membrane (Eadie and Kwan, 2008).
Pharmacokinetics
Representative values for the more important pharmacokinetic parameters of phenobarbitone and its congeners following initial doses of the agents are shown in table 1. Taken orally, these barbiturate derivatives appear to be fully or almost fully bioavailable. The older belief that methylphenobarbitone was only 50% bioavailable arose from a failure to appreciate that the drug was extensively transformed to phenolic metabolites as well as to phenobarbitone. The agents are eliminated mainly by metabolism (figure 1), involving oxidations initially, followed by conjugation of the first stage metabolites and, in the case of phenobarbitone, also by direct N-glucosidation. The long elimination half-life of phenobarbitone (3-4 days), compared with those of almost all other antiepileptic drugs, necessitates waiting two or three weeks after each dosage change before the full effects of the change are present. Phenobarbitone's slow elimination means a once daily dosage can satisfactorily maintain stable drug concentrations in biological fluids. While the long half-life slows the process of dosage adjustment for phenobarbitone and its congeners, it also diminishes the chance of seizures recurring, at least in adults, if there are short periods, e.g. a day or two, during which time drug intake is incomplete.
Phenobarbitone clearance increases in pregnancy.
Clinical use
Phenobarbitone (and its congeners) is effective in controlling focal seizures, regardless of whether they become secondary generalised, as well as myoclonic and convulsive seizures of idiopathic generalised epilepsies. Parental dosage forms are used to treat convulsive status epilepticus (Fincham and Schottelius, 2002).
Satisfactory oral phenobarbitone maintenance dosage in adults is likely to be around 1.0-2.5 mg per kilogram per day. However, it is often prudent to commence with lower doses to avoid initial sedation. Dosage on a bodyweight basis is likely to be higher in children, though lower in the very young and the elderly.
Methylphenobarbitone doses, mg for mg, are nearly twice those of phenobarbitone. Because the first primidone dose very occasionally causes prolonged sleepiness, it is wise to commence this drug with a very low dose, for example 62.5 mg or 125 mg, to see whether there are problems before increasing the dose. The average adult dose is around 10 mg per kg per day.
Steady-state plasma phenobarbitone levels in the range of 45 to 130 μmol per litre often correlate with anti-seizure effects of the drug, and of its congeners, without prohibitive adverse effects.
Interactions
Phenobarbitone is a well-established inducer of the cytochrome P450 system of enzymes, and the associated UDP-glucuronosyl transferases. These inductions may increase the clearance of certain endogenous molecules and numerous drugs which are broken down by these pathways if they are co-administered. Conversely, co-administered drugs that are metabolised by, or inhibit, these pathways may alter plasma phenobarbitone concentrations, either lowering or raising them depending on the drug in question. In particular, valproic acid may cause a substantial rise in plasma phenobarbitone concentration through competition for glucose conjugation capacity. More detailed accounts of the interactions are available elsewhere (Patsalos and Perucca, 2003a; Patsalos and Perucca, 2003b).
Adverse effects
Phenobarbitone and its congeners have dose-related sedative effects which can prove unacceptable in significant numbers of patients. These effects sometimes occur at concentrations that would be regarded as sub-therapeutic. Sedation may sometimes not be overt but cause headaches, mood alterations (mainly depression), and, in children, behavioural and cognitive disturbance and deterioration in school performance. Paradoxically, children may become hyperactive when taking the drug. At higher degrees of overdosage, nystagmus and ataxia of gait may occur.
Because phenobarbitone induces the hepatic cytochrome P450 system, its prolonged use may lead to a state of relative vitamin D deficiency and possibly osteomalacia.
Certain data suggest that phenobarbitone may be a human teratogen, though there is some uncertainty in this regard.
Phenytoin
Introduction
The chemical structure of phenytoin is related to that of barbiturates. Its synthesis was reported by 1908, followed by a report that it elevated the threshold of electrically induced seizures in cats. Subsequently, it was shown to provide effective treatment for epilepsy without causing sedation, a major advantage over barbiturates. Since that time, it has been one of the mainstays of seizure therapy. Its efficacy in treating epileptic status, when given intravenously, was demonstrated in 1968 (Wallis et al., 1968).
Phenytoin is marketed for oral administration and intravenous use.
Mechanism of action
It is likely that the main mechanism of action of phenytoin is to block frequency, use and voltage-dependent neuronal sodium channels, thus limiting the repetitive firing of action potentials. At therapeutic levels, there is little change in normal patterns of firing. At higher levels, phenytoin can impair the function of healthy neurons.
Pharmacokinetics
The absorption rate varies with different formulations, with peak levels appearing between four and eight hours after ingestion. Its bioavailabilty is in the order of 90%. Calcium sulphate added to the formulation, and antacids, retard absorption. Little is absorbed from the rectum. Intramuscular administration results in erratic absorption.
Distribution is rapid, with maximum concentrations being reached in the brain 15 minutes after intravenous administration. Its concentration in CSF usually equals the phenytoin unbound concentration in plasma, but may be slightly higher in saliva. Phenytoin passes into the placenta. Its concentrations in the umbilical cord are the same as those in the mother's plasma. The apparent volume of distribution ranges from 0.5 to 0.8 L/kg in adults and 0.8 to 1.2 L/kg in children. Phenytoin is about 90% bound to plasma protein. The proportion bound is lower in patients with poor renal function, with low albumin levels, and in pregnancy. Other drugs, e.g. salycilates and valproate, may reduce protein binding of phenytoin by competing for binding sites.
Elimination
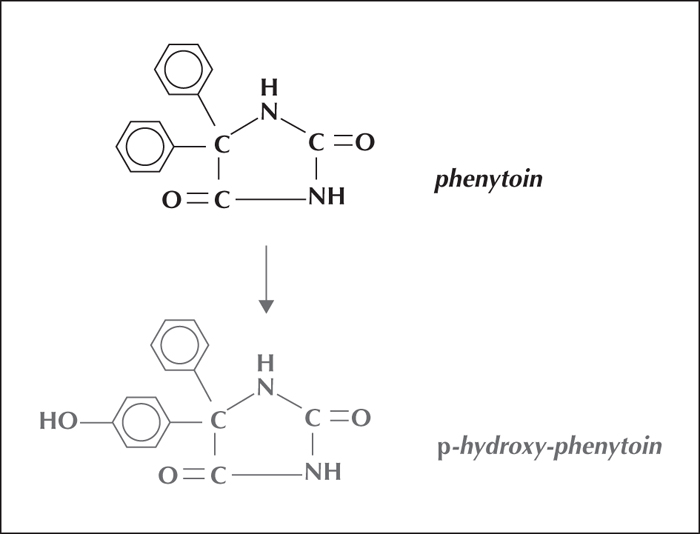
Phenytoin is eliminated almost exclusively by extensive metabolic hydroxylation via an arene oxidation product (figure 2). The para-(4)-hydroxylation is mediated largely by the cytochrome P450C2 subfamily isoenzymes, with CYP2C9 handling the major portion of the dose and CYP2C19 the rest (10%).
The capacity to metabolise phenytoin varies widely between individuals, largely because of genetic factors.
Clearance
Clearance values range widely from 0.015 to 0.065 L/kg/h, the higher values occurring in children. Plasma half-life values of 8-15 hours are found after a 100-mg test dose, but following overdose the half-life value may be as long as several days. Saturation elimination kinetics exist and result in disproportionate rises in plasma levels, relative to dose increases, once a limiting ceiling has been reached (Kutt and Harden, 1999).
Plasma levels in relation to clinical response
The majority of patients show a reasonable correlation between plasma levels and efficacy, while higher plasma levels are associated with toxicity. More so than other antiepileptic drugs, phenytoin plasma levels have a clinical application as a guide to management. The optimal range lies between 10 and 20 mcg/mL. It may be worth stressing that some patients, particularly those with newly-diagnosed epilepsy, respond to levels below this range.
Clinical use
Phenytoin is effective in controlling convulsive seizures, both in idiopathic generalised epilepsy and in focal epilepsy with secondarily generalised tonic-clonic seizures. It does not control any of the non-convulsive manifestations of generalised epilepsy and is not indicated in symptomatic generalised epilepsy, such as Lennox-Gastaut syndrome. Like most antiepileptic drugs, phenytoin ought to be introduced slowly and doses increased gradually until the control of seizures is achieved. This process can be guided by plasma level measurements, which are only an adjunct to the clinical response. Conventional doses in adults are in the order of 200-400 mg daily. The recommended initial dose range for children is in the order of 5 mg/kg/day, the maintenance dose usually being 4-8 mg per/day (maximum 300 mg per day), but individually variable (Stern et al., 2008).
Interactions
Phenytoin is a P450 enzyme inducer. Interactions with other drugs, both antiepileptic and other concomitantly administered classes of drugs, are common. Significant interactions are with drugs which cause a rise in phenytoin levels, including some antibiotics, benzodiazepines, warfarin, and many others.
Phenytoin reduces the plasma level of substances, including sex hormones, steroids, some antibiotics, psychotropic drugs, and lamotrigine. Its effects on the cytochrome enzymes may cause a lowering of vitamin D metabolites and thus contribute to metabolic bone disease, as well as affecting cholesterol metabolism. More details of the interactions with phenytoin are available elsewhere (Patsalos and Perucca, 2003a; Patsalos and Perucca, 2003b).
Adverse effects
The central nervous system effects of nystagmus, incoordination, and ataxia are dose-related. Somnolence and mood changes, nausea, extrapyramidal abnormalities, cerebello-vestibular toxicity, and mental changes have been reported. High dosages may cause seizure aggravation.
Skin rashes, cosmetic facial and gum-related changes, hypersensitivity, and lymphoma, which is not always benign, may occur. Occasionally, hepatotoxicity and, rarely, bone marrow abnormalities, including megaloblastic anaemia and a tendency towards bleeding, may occur. Decline of serum folate levels, low vitamin D levels, osteomalacia, and increased bone fractures have been widely reported. Intravenous phenytoin must be given slowly, should not mixed with other drugs, and may cause significant tissue irritation and discolouration and pain in the limb into which it is administered. Purple glove syndrome is also associated with oedema, and can be serious (Treiman, 1987; O’Brien et al., 2001).
Phenytoin has an application as a cardiac antiarrhythmic, but may also cause hypotension, arrhythmias, and collapse, if given rapidly.
Use in pregnancy and teratogenesis
Phenytoin is associated with a slightly increased incidence of foetal malformations. It is less teratogenic than valproate, but more teratogenic than lamotrigine. Cognitive changes in exposed offspring have not been proven. Phenytoin has been reported to interfere with male fertility.
Ethosuximide
In the mid-20th century, several succinimide derivatives were marketed. The only survivor is ethosuximide, which is effective in treating absence seizures of idiopathic generalised epilepsy. The drug is marketed in solid dosage forms and as solutions for oral intake.
Mechanism of action
Ethosuximide blocks thalamic ’T’ type calcium channels, an effect possessed by drugs that are effective against absence seizures.
Pharmacokinetics
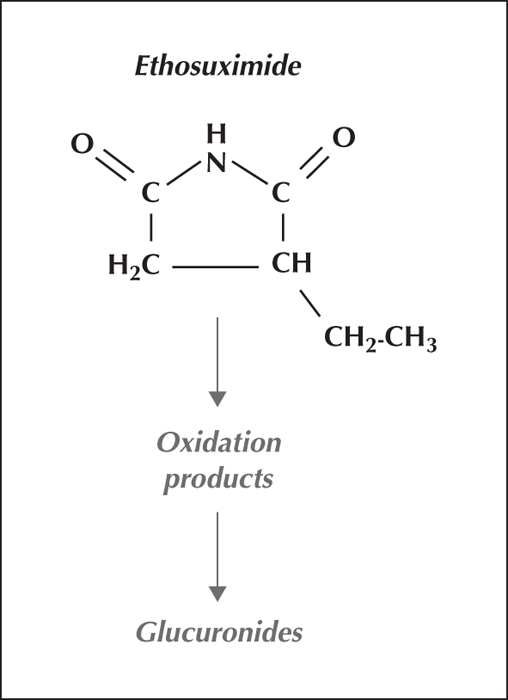
Ethosuximide is fully bioavailable orally, is not plasma protein bound, and is cleared by biotransformation to pharmacologically inactive metabolites (figure 3). During continued intake, its elimination half-life (around 30 hours) is long enough for a once-daily intake to maintain stable biological fluid concentrations over a 24-hour dosage interval.
Clinical use
Currently, ethosuximide is used mainly to treat absence seizures when more effective agents, in particular valproic acid, fail or are not tolerated. In the past, when ethosuximide was the mainstay of absence seizure therapy, it was suspected, whether justifiably or not, to sometimes precipitate the occurrence of convulsive seizures if used alone. Therefore, an antiepileptic drug effective against such seizures was often prescribed concurrently. Because absence seizures usually occur relatively frequently, ethosuximide dosage can often be adjusted satisfactorily in relation to the clinical response, after allowing for time to achieve steady-state effects after dosage changes (around 10 days). In theory, ethosuximide could be taken once daily, but the physical size and number of individual 250-mg dosage units required often makes divided daily dosage preferable. The usual therapeutic dose is around 30 mg per kilogram per day, and is likely to be associated with plasma ethosuximide levels of 300 to 700 μmol per litre (Bromfield, 2008; Glauser and Perucca, 2009).
Interactions
Ethosuximide exhibits relatively few drug-drug interactions. Further details are available elsewhere (Patsalos and Perucca, 2003a; Patsalos and Perucca, 2003b).
Adverse effects
Skin rashes and allergic and hypersensitive reactions are not common. Overdosage may cause sedation and altered behaviour. Little information is available regarding the drug's teratogenicity.
Carbamazepine
Carbamazepine was introduced to therapeutics as an antiepileptic agent and treatment for trigeminal neuralgia in the early 1960s. It has become the agent of first choice for focal epilepsies, and has some use in psychiatry and pain medicine. It is not effective in treating absence and myoclonic seizures of idiopathic generalised epilepsy (Silaanpaa et al., 2009).
The drug is marketed in solid and liquid dosage forms for oral use. Its low aqueous solubility has prevented satisfactory parental preparations from being developed. Modified release forms for oral use are available.
Mechanism of action
The main molecular mechanism of antiepileptic action of carbamazepine involves blocking voltage-dependent sodium ion channels in cell membranes.
Pharmacokinetics
Typical values for the major pharmacokinetic parameters of carbamazepine after initial administration are shown in table 1. The drug induces its own metabolism; after each dosage increase, its clearance increases and its plasma half-life shortens. The drug's elimination parameters during chronic intake may therefore differ from those shown in the table. Currently marketed carbamazepine preparations seem to have satisfactory and nearly complete or complete oral bioavailabilities, however, in the past, preparations were marketed with a particle size small enough to produce rapid rises in plasma drug concentration, resulting in temporary nystagmus and ataxia of gait after each dose. Generic manufacturers seem to have difficulty producing carbamazepine preparations with particle sizes and absorption profiles identical to those of the market leader. Therefore, it is sometimes recommended that patients always use the same brand of the drug after a satisfactory therapeutic regimen has been established (Eadie and Vajda, 1999).
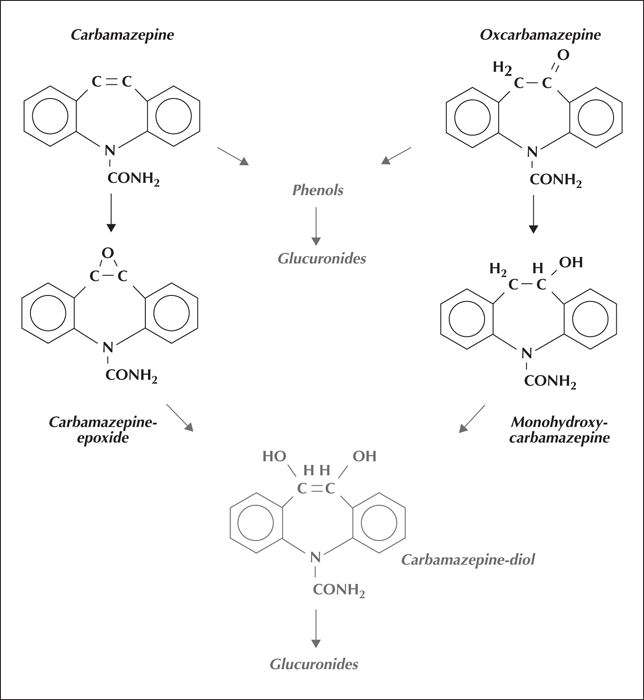
In the body, carbamazepine is converted to a biologically active epoxide metabolite (figure 4). This has a shorter half-life than carbamazepine, thus, in continued use, its plasma concentrations tend to parallel those of the parent substance. The epoxide is hydrolysed to a biologically inactive diol derivative, excreted mainly as a glucuronide conjugate (figure 4) (Dickinson et al., 1999).
Clinical use
Carbamazepine is currently considered the drug of first choice for managing focal epilepsies. The usual dose for continued use in adults and children is around 10-20 mg per kilogram per day, but may need to be higher. However, it is usually prudent to commence treatment with no more than half the expected dose over the next three or four weeks, perhaps guided by plasma carbamazepine concentrations, to increase the dose, as clinically indicated, to its expected value. The drug's continuing induction of its own metabolism over this period is likely to ensure that its concentrations in biological fluids remain relatively steady, despite the dosage increments. Unless seizure control has already been obtained, adjusting the dose to achieve steady-state plasma carbamazepine concentrations in the range 25 to 50 μmol per litre often offers the best chance of achieving seizure control without overdosage manifestations. The measurement of simultaneous plasma carbamazepine epoxide levels does not seem to offer a clinical advantage (Marson et al., 2007).
Interactions
Carbamazepine not only induces the capacity of cytochrome P450 3A4 to enhance its own metabolism, but increases this enzyme system's capacity to metabolise endogenous molecules and numerous drugs that are biotransformed through this mechanism. Drugs that inhibit the enzyme system, for example isoniazid and erythromycin, if co-administered, will raise plasma carbamazepine levels. Further details of the known interactions are available elsewhere (Patsalos and Perucca, 2003a; Patsalos and Perucca, 2003b).
Adverse effects
Skin rashes occur soon after carbamazepine intake in some 5% or more patients. If drug intake is not ceased promptly, serious systemic toxicity may follow with, for instance, hepatitis. In overdosage, carbamazepine causes nystagmus and blurred vision, ataxia of gait, and drowsiness. Symptomatic hyponatraemia may occur occasionally and, if unrecognised, may impair seizure control and result in the prescription of higher carbamazepine dosages, thus worsening the problem. Significant bradycardia and heart block have been reported occasionally and asymptomatic neutropenia was noted with some frequency when the drug first became available. The induction of cytochrome P450 activity may produce a degree of vitamin D deficiency with possible osteomalacia.
There is little evidence that carbamazepine is a significant human teratogen, but a dose-related effect has been demonstrated (Fertig and Mattson, 2008).
Oxcarbazepine
Oxcarbazepine, a molecule similar to carbamazepine, has been marketed (in solid dosage forms) as an oral antiepileptic agent in many countries, but only in fairly recent times, although in some places it has been available for much longer. Its clinical development appears to have suffered, possibly because the possible advantages over carbamazepine took time to explore (Beydoun et al., 2008).
Oxcarbazepine appears to have a similar mechanism of action to carbamazepine. It and carbamazepine share the same second-stage biologically-inactive diol metabolite. The initial stage of metabolism of oxcarbazepine yields a pharmacologically-active mono-hydroxy derivative, not formed by oxidation, thus cytochrome P450 activity is not involved. Consequently, oxcarbazepine is less prone to drug-drug metabolic interactions than carbamazepine (Faught and Limdi, 2009).
Oxcarbazepine itself is eliminated very rapidly (figure 4) and in effect serves as a prodrug for its mono-hydroxy metabolite (with a half-life of around 10 hours). Clinically, 300 mg of oxcarbazepine seems to have similar therapeutic effects to 200 mg of carbamazepine. On the basis of this equivalent dose, oxcarbazepine seems less sedating than carbamazepine, but more prone to causing hyponatraemia. Its lower sedative tendency may allow it to be used instead of carbamazepine when maximum tolerated doses of the latter have reduced seizure frequency but not prevented all seizures.
Sodium valproate (valproic acid, VPA)
Originally used as a solvent for barbiturate hypnotics, valproic acid was accidentally discovered to be more effective as an antiepileptic than the test drug. Introduced in France in the 1960s, it rapidly became the drug of choice for generalised epilepsies. It is used also for treating psychiatric illness, mainly bipolar disorders, and for migraine prevention. Sustained-release formulations exist.
Chemistry
Valproate is n-propyl pentanoate (di-n-propyl acetate), a branched chain fatty acid. It is marketed in free acid form or the corresponding sodium salt, and also as divalproex sodium, a combination of valproate and valproic acid. An intravenous formulation is available. The rectally administered drug has been reported to be effective.
Animal studies
Valproate suppresses the spread of seizure activity, but is not effective in inhibiting focal discharges. It has no effect on focal electrical after-discharges or on kindled focal seizures.
Mechanism of action
Its mechanism of action is complex. Multiple mechanisms have been reported, including elevation of brain GABAergic, inhibitory activity. It decreases cortical excitability and affects a variety of brain receptors and limits high-frequency repetitive firing of Na+-dependent action potentials, through blockade of Na+ channels.
Clinical use
The drug has a wide spectrum of activity and is the agent of first choice for idiopathic generalised epilepsy, being effective against all manifestations of the disorder, including absences. It has been used synergistically with ethosuximide, and with lamotrigine. It is effective against focal seizures, but perhaps less so than carbamazepine, and is reported to be effective against febrile seizure prophylaxis and Lennox-Gastaut syndrome, although the evidence is not conclusive and it should not be used routinely for these indications.
Valproate should be introduced slowly and gradually until effective seizure control is established, rather than in standard doses of 1,000-1,500 mg per day in adults, as traditionally used, when it is more likely to be associated with unwanted effects.
The drug is highly effective for the treatment of juvenile myoclonic epilepsy, although the treatment may need to be continued indefinitely.
In pregnancy, it is teratogenic in a dose-dependent manner, more so than any other currently widely-used antiepileptic agent. If it must be prescribed in pregnant women, it should be used in the lowest effective dose, especially in the first trimester. In addition to physical malformations, valproate intake during pregnancy may be associated with cognitive impairment in the offspring (Vajda et al., 2010). Alternative agents are often not as effective as valproate. In an established pregnancy, replacement of valproate medication is likely to have problematic consequences (Jentink et al., 2010; Tomson et al., 2011).
Plasma level-clinical effect relationships
The population-based therapeutic range of 350-700 micromol/L is only a rough adjunctive guide to efficacy; the measurements are most useful in assessing compliance. Its biological effects and plasma levels are not closely correlated (Vajda, 2000).
Pharmacokinetics
All formulations are absorbed completely. The enteric-coated formulation is absorbed slowly, with a 2-3-hour time lag. Peak levels from enteric-coated tablets occur 3-5 hours after intake. Plasma concentrations after rectal administration have been comparable to those after oral intake. Valproate has a small volume of distribution due to high affinity for plasma albumin. A free fraction value of 10% was reported, increasing at higher total valproate concentrations. Lower protein binding has been noted in women in late pregnancy and the elderly, and in liver and renal disease.
Valproate is highly lipophilic and highly concentrated in the liver. It enters the brain rapidly, reflected by its prompt antiepileptic effect (Shen and Levy, 1999).
Clearance is increased at high doses, due to the higher free fraction of valproate. The clearance is notably higher in patients receiving CYP450 enzyme inducers. Neonates have a low and immature microsomal drug metabolising system, but eliminate valproate at the same rate as adults, despite having a larger circulating free fraction. In adult life, clearance of valproate appears to remain stable. Intrinsic clearance is lower in the elderly. Dosing requirements are generally guided by unbound clearance, not by total clearance, and unbound valproic acid clearance is not increased at higher doses.
Valproate is extensively metabolised, through oxidative and conjugation mechanisms (figure 5). Conjugation with D-glucuronic acid represents the major pathway of biotransformation, accounting for 30-40% of the dose. Similar to fatty acids, valproate is sequentially oxidised by mitochondrial beta oxidation, to yield 3-oxo-valproate. Other pathways are also involved, yielding numerous metabolites. Omega and omega-1 oxidative pathways have been described. The metabolites appear to be biologically inactive.
Interactions
Phenytoin, carbamazepine, and phenobarbitone cause a twofold increase in valproate clearance due to their effect on UDP-glucuronosyl transferase, and a less marked effect on cytochrome P450-mediated pathways. Valproate affects four enzyme systems involved in the elimination of many drugs, but many of the interactions are not important clinically. Lamotrigine elimination is inhibited, but there is also a synergistic pharmacodynamic interaction with clinical benefits when these two agents are combined.
Adverse effects
Gastrointestinal effects, hair loss, and weight gain, have been reported, but much more important is the finding of teratogenicity associated with valproate which is dose-dependent and may be associated with severe birth defects, such as spina bifida, cardiac abnormalities, skeletal, and renal malformations, as well as a decline in cognition, IQ, language, and possible autism (Meador et al., 2009).
Valproate may contribute to the development of polycystic ovaries and to polycystic ovarian syndrome. This is likely to be multifactorial in causation, involving genetic factors, diet, and the use of valproate in monotherapy rather than polytherapy (Isojarvi et al., 1998; Genton et al., 2001).
Valproate has also been reported to cause reduced sperm function. Hepatic failure and pancreatic necrosis have been reported in 1:15,000 patients, usually occurring in severely ill babies and young children. In contrast, liver function test elevations of no clinical significance occur in about 60% of patients treated with antiepileptic drugs. The risk of severe liver toxicity can be much greater in certain risk groups, particularly babies (e.g. with genetic metabolic defects).
Benzodiazepines
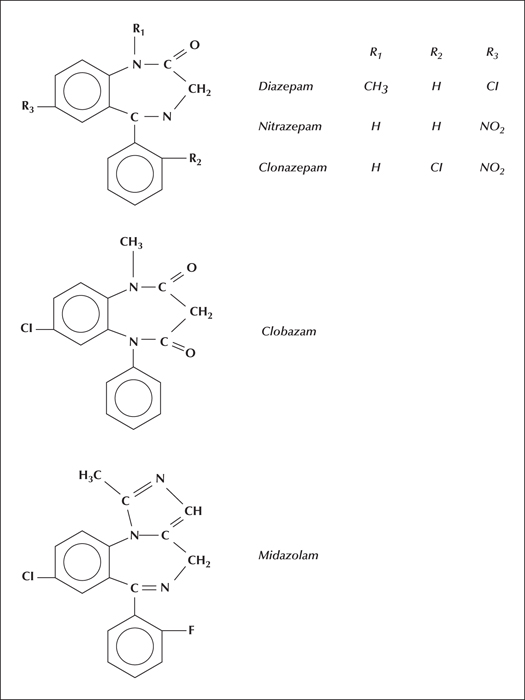
Benzodiazepines are used as antiepileptic drugs mainly in particular situations, such as status epilepticus (diazepam and clonazepam), in contingency situations (clobazam), and in some paediatric syndromes (nitrazepam). These will be discussed as a group and as individual substances (Panayiotopolous, 2005).
Animal models
Diazepam, clonazepam, nitrazepam, and clobazam are four members of the group which are effective in elevating the threshold for pentylenetetrazole-induced seizures. These drugs are also effective against maximal electroshock-related seizures. In chemically induced seizures, they can suppress the spread of electrical discharges (Shen and Levy, 1999).
Mechanisms of action
Benzodiazepines act by facilitating the synaptic action of the inhibitory neurotransmitter GABA (gamma amino butyric acid). They have no effect on synapses which do not involve GABA. The GABAA receptor is specific to benzodiazepines. Binding increases the frequency with which chloride channels are opened, increasing inhibitory neurotransmission. Benzodiazepines also have an effect on sodium channels and reduce voltage-dependent calcium currents, although their profile is different from the clinical effects of classic sodium and calcium channel blockers (Macdonald, 2003).
Adverse effects
Adverse effects are numerous, including neurological and psychiatric effects, sedation, memory problems, hyperactivity, withdrawal effects, and tolerance. Drowsiness is more common with drugs with long half-lives. Tolerance appears more rapid with drugs with short half-lives. Rebound insomnia may occur after withdrawal (Schmidt and Wilensky, 2008).
Individual benzodiazepines
The structure of the benzodiazepines discussed individually below is presented in figure 6 (Eadie and Vajda, 1999).
Diazepam
Diazepam was the first benzodiazepine used in epilepsy and its effectiveness depends on rapid absorption, distribution, and brain penetration. It binds to specific GABA receptors. It is sedative, and it is difficult to maintain its concentration, hence tolerance develops. Diazepam is unsuitable for long-term management of seizure disorders.
Pharmacokinetics
The drug is rapidly absorbed and mean peak plasma levels are obtained within 30-90 minutes after oral administration. Intramuscular administration is better avoided due to poor and irregular absorption. The rectal route leads to satisfactory absorption, although less predictably than the oral route, diazepam is rapidly and widely distributed, crossing the blood/brain barrier rapidly. Diazepam concentrations in the CSF are related to its unbound fraction in plasma. Rapid concentration peaks occurring in the grey matter correlate with a rapid onset of biological action (Kilpatrick, 1999).
Diazepam is highly protein-bound, independently of its total concentration in plasma.
Diazepam is eliminated by biotransformation in the liver (desmethylation and oxidation). N-desmethyldiazepam itself is a pharmacologically-active metabolite. Excretion takes place in the kidneys. Little unchanged diazepam is found in human urine, the dose being largely metabolised. The metabolites are excreted after conjugation with glucuronic acid.
No correlation has been found between plasma levels of diazepam and clinical effects.
Clinical effects
Status epilepticus: Following a single intravenous injection of 10-20 mg in adults and 0.2-0.3 mg/kg in children, given at a rate of 2-5 mg/min, brain concentration of the drug will be attained that is adequate for an acute antiepileptic effect. This effect may be lost after re-distribution, such that repeated bolus doses may be required to maintain brain drug concentrations. Higher doses may cause narcosis, hypotension, and respiratory arrest.
In practice, a benzoidiazepine drug ought to be used as first-line treatment for status epilepticus, supplemented from the outset by a major, chronically-effective antiepileptic drug.
Interactions
An interaction with phenytoin has been reported, resulting in high plasma levels of phenytoin, associated with symptoms of phenytoin intoxication.
Clonazepam
Clonazepam is used as an adjuvant in antiepileptic therapy, and for the treatment of status epilepticus. It has been used for a variety of epileptic syndromes, such as West and Lennox-Gastaut syndromes.
Although not as likely to induce tolerance as clobazam, clonazapam is more likely to lead to seizures if withdrawn rapidly. It has found an extensive application in psychiatry.
Clonazepam is well absorbed, with peak plasma levels being obtained four hours after intake. Its distribution is rapid and its apparent volume of distribution is in the order 1.5-4.4 L/kg. Protein binding in plasma is in the order of 85%. It is extensively metabolised, producing no active metabolites. Its elimination half-life is 22-33 hours, and is similar after oral and intravenous administration.
Plasma clonazepam concentrations vary widely between individuals. There is no correlation between plasma concentrations and therapeutic effects.
Adverse effects
Drowsiness and incoordination, habituation, and personality changes have been described.
Clobazam
This drug is a 1,5 benzodiazepine derivative, in contrast to clonazepam which is a 1,4 benzodiazepine. Clobazam is relatively insoluble. No intravenous preparation exists.
Clobazam and its metabolite N-methyldesmethylclobazam are both antiepileptic in a variety of seizure models and enhance GABA-activated currents. The chronic effectiveness of clobazam may be predominantly due to its active metabolite.
Clobazam is effective across a broad spectrum of epilepsy syndromes, both as monotherapy and as adjunctive therapy. Although doses up to 60-80 mg per day have been used in adults, it is generally accepted that the development of tolerance precludes the use of doses over 35 mg per day. Many respond to lower doses. Tolerance may occur within weeks of starting treatment, but it is uncommon at doses of 10-30 mg per day. The drug can be used effectively in contingency situations, such as a high level of stress, sleep deprivation, or aeroplane travel (Loscher and Schmidt, 2006).
Pharmacokinetics
Over 80% of the dose is absorbed. Rectal administration has been reported to be effective. Its distribution volume increases with age. Salivary concentrations correspond to plasma concentrations. The drug is over 80% protein bound, and is eliminated by metabolism, the main pathway involving demethylation and hydroxylation. Its elimination half-life is in the order of 10-15 hours, although longer in the elderly.
A relationship between plasma level and clinical effects has not been demonstrated. Clobazam may increase the plasma concentration of co-administered phenytoin, valproate, and phenobarbitone.
Adverse effects
These are mild. Drowsiness and fatigue tend to be transient.
Nitrazepam
This drug is essentially a hypnotic. It is well absorbed and peak levels occur after two hours. Its elimination half-life is 20-36 hours. It is used only for intractable seizures associated with symptomatic epilepsies in childhood.
Midazolam
This drug is water soluble and can be given intramuscularly. It is suitable for use in status epilepticus, rapidly achieving high plasma concentrations. It has a brief elimination half-life of about three hours.
Midazolam provides less sedation, which is a decided advantage in status epilepticus. Used intramuscularly, it is a useful alternative to intravenous clonazepam or diazepam.
In some countries, lorazepam, another benzodiazepine, is the drug of choice for the treatment of status epilepticus.
Disclosures
The authors have no conflicts of interest to disclose.