Epileptic Disorders
MENUEpileptic spasms in paediatric post-traumatic epilepsy at a tertiary referral centre Volume 19, numéro 1, March 2017

Post-traumatic epilepsy (PTE) in children after a traumatic brain injury (TBI) occurs in 10-20% of children following severe head trauma (Ratan et al., 1999; Barlow et al., 2000; Appleton and Demellweek, 2002). The presence of two or more unprovoked seizures following TBI defines PTE (Annegers et al., 1980). Epileptic spasms (ES) may occur after a cerebral injury in infants and children, and are characterized by a sudden, symmetric or asymmetric flexion or extension of the axial and/or proximal limbs (Kellaway et al., 1979; Egli et al., 1985; Scantlebury et al., 2010). This seizure type occurs primarily in infants and is generally associated with a poor long-term outcome. ES, however, may persist or appear beyond the first year of life and even during late childhood (Riikonen, 1982; Bednarek et al., 1998). Aetiologies for the development of ES include genetic, metabolic, and symptomatic causes but the pathophysiology of ES remains unclear despite the development of various animal models (Scantlebury et al., 2010).
The incidence rate of ES in PTE in children is unknown. In this study, we analysed clinical variables in a subset of 47 patients with PTE, all evaluated at a tertiary paediatric referral centre; all subjects developed epilepsy after either an accidental or non-accidental head injury. Since ES following TBI is an acquired form of ES in a previously normal child, we hypothesized that understanding its epidemiological and clinical characteristics may provide some insight into mechanisms of epileptogenesis in ES.
Study design and methods
Patient identification
A retrospective electronic chart review was performed at the Children's Hospital of Michigan (CHM) from 2002 to 2012. Both inpatient and outpatient charts were screened for patients who were referred after a TBI to any one of the seven child neurologists. These patients had diagnoses related to their TBI including post-concussive syndrome, seizures, and focal neurological deficit(s). The following clinical variables were collected: demographics, age at TBI, mechanism of trauma, severity of TBI (mild, moderate, or severe), neurological surgery interventions, seizure semiology, perinatal complications, type of trauma, i.e. accidental (AT) and non-accidental (NAT), imaging and neurophysiological data, onset of first seizure/spasms, treatment/response, comorbidities, follow-up duration, and outcome.
Seizure onset time, as published by Iudice and Murri (2000), was classified as immediate, early, or late; immediate (8 days after injury). Severity of TBI was classified as mild for loss of consciousness or amnesia for less than 30 minutes; moderate for loss of consciousness from 30 minutes to one day; and severe for loss of consciousness of more than one day, presence of parenchymal haemorrhage, subdural haematoma (SDH), skull fracture, and/or brain contusion (Annegers and Coan, 2000). Parenchymal haemorrhage and skull fracture were included as features of severe injury in our study.
Inclusion and exclusion criteria
Only the patients referred to a child neurologist, both inpatients and outpatients, were included in the study. Typically, outpatient referrals after TBI included evaluations for symptoms of concussion, seizure, and other related acute and/or chronic comorbidities. This cohort also included those who were seen in the hospital for consultation after TBI. Inpatient referrals were for specific indication(s) related to the head injury, including a pre-/in-hospital paroxysmal event after a TBI. The referring physicians were primary care physicians, hospitalists, intensivists, neurosurgeons, or physiatrists. Not all patients hospitalized after a head injury were seen by a child neurologist unless a consultation was requested. Only the new cases of PTE during the 10-year period were investigated during the study. For example, if a child was diagnosed with PTE in 2003, this patient was followed until 2012 or until the patient was lost to follow-up sooner. After a retrospective chart review of 321 patients who suffered TBI and were followed for a minimum of nine months and maximum of eight years and five months, 47 were noted to have developed PTE. Patients who only had one seizure within the first week of head injury and without any additional unprovoked seizures before the cut-off time period were excluded. All child neurologists in our department are board-certified and had equal participation in evaluating patients. All patients seen in both inpatient and outpatient settings were followed at varying times yearly in the clinics for the development of PTE. Diagnosis of epilepsy was invariably confirmed with EEG and/or video-EEG during the initial or subsequent evaluation(s). This study excluded patients who had subclinical seizures in the paediatric intensive care unit, neonates with hypoxic ischaemic injury at birth, or patients with history of provoked or unprovoked seizure prior to TBI.
Seizure classification and diagnosis
The semiological classification of seizures used was based on history, and when available, video-EEG data (Lüders et al., 1998). This classification allowed identification of ictal features and categorization of seizures based on the degree of information available about the seizures. All seizure histories were obtained through interviews with a seizure witness, or witnesses, if possible and/or patient, if able. Subsequent seizure histories were well documented by the neurologist in subsequent follow-up clinic visits, accompanied by results of outpatient EEG and/or inpatient video-EEG. It is possible that some patients may have had subtle clinical seizures that may have been missed by the caretaker, but we conclude that this is insignificant since patients with PTE in our cohort presented with numerous clear seizures.
Statistical analysis
Continuously scaled variables were evaluated using medians and ranges. Categorically scaled variables were analysed using proportions and ratios. Comparisons between cohorts on categorically scaled variables were obtained using a non-parametric Fisher's exact Chi-square test. The Wilcox Rank sums test was used to compare groups on continuously scaled variables. The SAS System software® was used for statistical analysis. Statically significant difference was considered present at a p value
There were two patients with epileptic spasms who were aged less than one month at the time of TBI and one patient with an unknown number of current AEDs. For statistical purposes, the former two patients were assumed to be 0.3 months of age, by assuming age at injury to be approximately one week old.
Results
Characteristics of the spasm cohort
The characteristics of the overall cohort have been published previously (Park and Chugani, 2015). Out of the 321 patients who had TBI, 47 (15%) patients were diagnosed with PTE. Of the eight patients (out of 47) who suffered mild TBI, seven children were between 3 and 11 years of age at the time of injury, and one was 23 months old. All infants (table 1 table 1). There were two males and four females whose age at the time of injury ranged from less than one month to eight years. The age range of the patients who suffered TBI due to NAT was 2-3 months. This is in agreement with published data that most cases of inflicted head injury occur in children under one year of age (McClelland et al., 1980).Follow-up duration ranged from nine months to eight years and five months. Two patients, Patients 5 and 6, had neurosurgical intervention, i.e. shunts, for hydrocephalus.
Comparison of patients with “favourable” and “unfavourable” (ES) outcome (table 2 table 2)showed no significant differences with regards to gender, type of injury (NAT vs AT), or presence/absence of skull fracture. Significant differences (a higher chance of developing ES) were observed in patients with two or more seizure types, taking two or more AEDs.
Seizure semiology and EEG
In addition to ES, all six patients had other seizure type(s). Both focal and generalized seizure semiology (Patient 1) was seen in both groups of six patients, NAT vs AT. All patients with ES had multiple irritative zones manifesting as multifocal epileptiform discharges, unilateral or bilateral, independent of the mechanism of cerebral insult. The most common long-term interictal EEG finding was multifocal epileptiform discharges in bilateral hemispheres, except in Patient 3. EEG in Patient 1 evolved over eight months, from normal background with bifronto-central spikes and waves to generalized slow spike and waves and multifocal polyspikes. An electroclinical diagnosis of Lennox-Gastaut syndrome (LGS) was made. Routine EEG performed within 48 hours of injury in Patients 2 and 5 showed no epileptiform discharges, but evolved over 4-6 months to show multifocal epileptiform discharges.
Therapeutics and outcome
The ketogenic diet was used in one patient with limited success. Patient 1 who was in a motor vehicle accident at 8 years of age had a greater than 50% decrease in seizure frequency and decreased seizure intensity three months after initiation of the ketogenic diet, while remaining on multiple AEDs. She was initially seizure-free for about three weeks at the initiation of the ketogenic diet.
Albeit at various success rates, spasm freedom was achieved with either vigabatrin or ACTH. Patients 2-4 became spasm-free after treatment with vigabatrin as the initial treatment for spasms, although the other seizure types continued. Patients 5 and 6 were initially placed on vigabatrin, but only became seizure-free after the initiation of ACTH. For Patient 5, all AEDs were weaned off and the EEG, four years after the last seizure (at 5 years of age), showed 6-7-Hz posterior dominant rhythm with no epileptiform discharges. Patient 3 was evaluated for epilepsy surgery, but was not a candidate due to multifocal epilepsy. All patients were maintained on AED polytherapy, ranging from 2-5 medications. Five of the six patients had refractory epilepsy. Cognitive delay and epileptic encephalopathy were seen in all six patients, five of whom were free of spasms after treatment with vigabatrin or ACTH. Other comorbidities included cortical blindness, ataxia, spastic quadriplegia, hydrocephalus, and hemiparesis.
Neuroimaging
Computed tomography (CT) findings were available in five patients, four of whom had skull fracture acutely; one had subdural haemorrhage. Not all patients had brain magnetic resonance imaging (MRI). Patient 1, four weeks after TBI due to a motor vehicle accident, showed MRI findings in the corpus callosum suggestive of remote shearing injury. There was also volume loss, enlarged ventricles, and a diminished size of the left hippocampus and left portion of the brain stem. Patient 2 had acute skull fracture and acutely showed chronic multicystic encephalomalacia on MRI. Patient 4 showed MRI findings suggestive of diffuse hypoxic damage in the multiple regions of both hemispheres. The timing of brain MRI was not at all consistent, and ranged from weeks to months. Only one, Patient 3, had [18F] flurodeoxyglucose (FDG) positron emission tomography (PET) which showed varying degrees of decreased metabolism in the bilateral hemispheres.
Discussion
EEG
Observation of our small cohort demonstrates that ES may develop after TBI as the first seizure type or follow the onset of other seizure types. ES may also start months after resolution of previous seizure type(s), and in the absence of cortical irritation manifesting as interictal epileptiform discharges. Birca et al. (2014) reported two patients with NAT who had resolution of focal seizures lasting for months, one with absent epileptiform discharges during a period of seizure freedom for two months, before spasms developed. The evolving nature of the gliotic process and neuronal connections in the setting of an immature brain, after a severe cerebral insult, may partly explain the appearance of a new seizure type even in the absence of interictal epileptiform discharges over a period of months.
A series of new EEG findings in the context of increasing seizure severity in our cohort prompted specific therapeutic modifications. EEGs in Patients 1, 2 and 5 evolved over months, from the time of TBI, showing signs of increased cortical irritation. The interictal EEG in Patient 2 correlated with the brain MRI showing multicystic encephalomalacia, suggesting multiple sites of cerebral injury that translate to cortical irritation. Similarly, EEG in Patient 1 evolved from bifrontocentral epileptiform discharges to generalized and multifocal epileptiform discharges. These illustrate evolution of interictal epileptiform discharges in these patients with severe TBI and intractable epilepsy. Based on this observation, repeating a routine EEG, 3-6 months after the initial evaluation in these patients, may be beneficial in order to monitor for changes in the patient's status that may not be clinically obvious.
Monitoring for the development of ES needs to continue for at least two years post cerebral insult, even if the age at the time of the insult is beyond toddler age and the patients are already manifesting other seizure type(s). The shortest and longest intervals between the age at TBI and onset of ES in our cohort were two months and two years, seen in Patients 3 and 2, respectively. During the latent period preceding the onset of ES, a probable reorganization and an abnormal interaction between the injured cortex and subcortical regions/brainstem likely exists, and if a certain threshold of abnormal interaction is reached, spasms may start. Hypsarrhythmia may be the first EEG manifestation of this abnormal interaction, although this EEG finding is not necessary at the time of occurrence of epileptic spasms (Caraballo et al., 2003). Not all severe TBI in children result in ES, as shown in our previous study (Park and Chugani, 2015), thus yet another variable likely exists in the development of ES, such as genetic predisposition.
Therapeutic effect of vigabatrin vs ACTH
Vigabatrin was approved in the United States in 2009 for the treatment of ES in patients one month to 2 years of age (SABRIL Oral Solution, 2010). Overall, response rate in patients with various causes of ES, including tuberous sclerosis, is 36% (Elerman et al., 2001). In patients with TSC, the response rate was higher; 52% (Elerman et al., 2001), and over 90% in some studies (Thiele, 2004).
Our cohort all had multiple seizure types; one patient had ES as a seizure sequence following a generalized tonic seizure. Three patients’ ES resolved with vigabatrin and one patient had an unknown response, suggesting that there may be a response rate of at least 50-65% when treating ES in children with PTE and ES. This response rate is similar to that reported for TSC. However, we acknowledge that our sample size is too small to draw a meaningful conclusion about the efficacy. Two patients who failed vigabatrin responded to adrenocorticotropic hormone (ACTH), thus five of the six patients’ ES stopped on either vigabatrin or ACTH. It is unknown what the response rate is in patients with ES after trials of vigabatrin followed by ACTH or vice a versa after a TBI. Children with PTE and ES appear to show a promising response to one of the two medicines approved for the treatment of ES. Moreover, vigabatrin may be considered as the first line of therapy in this selected group of patients in order to avoid the side effects associated with ACTH.
Timely diagnosis of ES in this group of patients with PTE may positively impact cognitive outcome given that vigabatrin and ACTH have shown efficacy in treating epileptic spasms (Fejerman et al., 2000). However, it is unknown whether the resolution of spasms after treatment(s), with or without persistent other seizure type(s), improved cognitive impairment in this specific cohort who all suffered with severe TBI.
Focal vs generalized seizure semiology
The initial seizure type in all patients was difficult to lateralize, which may be in part due to the severity of TBI, with injuries to multiple regions in the brain. All (except Patient 1) were less than one year of age at the time of TBI. It is well known that focal seizures in infants can present with generalized seizure semiology, notably spasms or diffuse tonic seizures (Nordli, 2013). Moreover, infants who suffered severe TBI may be even more prone to generalized seizures from focal or multifocal sources. Both focal and generalized seizure semiologies (other than ES) were seen in both subgroups (NAT vs AT) of the six patients who developed ES. This observation suggests that even in these patients with an acquired head injury, whether there is a focal or multifocal insult, generalized seizure semiology can be seen.
There is controversy whether epileptic spasms are focal or generalized in nature. However, it is established that ES can be due to either a focal structural abnormality or metabolic/genetic disorder affecting the cortex “diffusely” (Chugani et al., 1990; Wyllie et al., 2007). Regardless of the cause of ES, an interaction between the abnormal cortex and the subcortical grey matter/brainstem appears to be necessary for the development of epileptic spasms in infancy (Frost and Hrachovy, 2005).
Latency of epileptic spasms
Delay in onset of epileptic spasms after TBI in the six patients ranged from two months to two years. This temporal delay between a cerebral insult and onset of ES has been reported by numerous investigators (Watanabe et al., 1973; Matsumoto et al., 1981; Velez et al., 1990; Okumura et al., 2001). Matsumoto et al. (1981), in a review of 200 cases of infantile spasms associated with various aetiologies, demonstrated a difference in age at onset of ES based on the time and nature of the initial cerebral insult. Prenatal or perinatal causes were associated with an earlier onset (0-3 months), cryptogenic causes an intermediate onset peak (4-6 months), and postnatal insult an even later onset of ES. This trend was not seen in our small “postnatal” subgroup. Five of the six patients suffered TBI from Day 1 of life to 2.5 months, but began having ES at two months, four months, seven months, one year, and two years, respectively, after the cerebral insult.
Other investigators suggested that the timing of the onset of ES is related to the location of the focal cortical lesions in different lobes that mature at different times (Koo and Hwang, 1996). They postulate that the cortical lesions in critical parts of the brain undergoing maturational changes may contribute to abnormal neuronal circuit interactions with the brainstem to generate spasms. Combining the neuroimaging and EEG data available for our six patients, it is most likely that all had injuries to multiple regions of the brain, thus we could not test this hypothesis that correlates the location of focal cortical lesions to the timing of the onset of ES.
Support for the hypothesis that injury to the frontal lobes may result in late onset of ES may be found in neuroimaging studies. The presence of progressive, regional brain maturation is supported by [18F] flurodeoxyglucose (FDG)-PET studies showing regional increases in glucose metabolism at different ages of CNS maturation (Chugani et al., 1987). Neuroimaging studies by Barkovich et al. (1988) also showed ontogenic changes of brain myelination on MRI with T1-weighted images in the developing infant and child. These studies indicate that the frontal lobes mature last. Indeed, it is now known that the brain is not completely mature until the early third decade (Hassink et al., 1992). Furthermore, in a study of late-onset epileptic spasms in 34 patients with a mean age at onset of spasms of 3.1 years, 76.2% of those with focal EEG abnormalities showed anteriorly predominant findings, possibly suggesting involvement of frontotemporal regions. Another study of late-onset epileptic spasms in 19 patients, with a median age at 3 years and 9 months, showed similar findings pointing to abnormal EEG in frontal, temporal, or frontotemporal regions (Bednarek et al., 1998; Eisermann et al., 2006). Late-onset epileptic spasms in our 8-year-old (Patient 1) may support this hypothesis since she had mostly bi-frontal lobe injury, as evidenced by her first EEG when her seizures (other than ES) began. The EEG showed bi-frontocentral spikes with normal posterior dominant rhythm for age, indicating regional irritation involving the frontal lobes. One may also speculate that the use of a sodium channel blocker (SCB), such as oxcarbazepine (OXC), before the onset of ES (although we are unable to verify the timing of initiation) may have had a role in ES onset (Veerapandiyan et al., 2012).
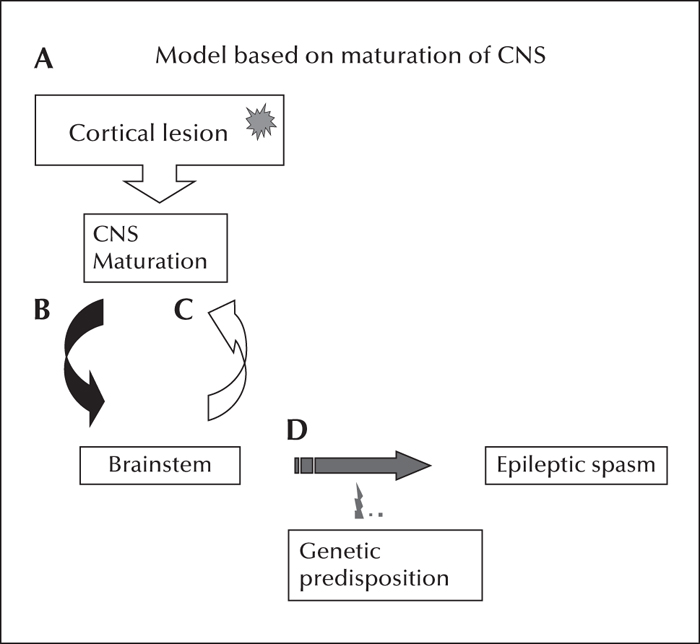
The temporal association between a cerebral insult and onset of epileptic spasms in PTE is also supported by the maturation time course of synaptogenesis in the human brain (Huttenlocher and De Courten, 1987; Huttenlocher and Dabholkar, 1997). There is a rapid increase in synaptic density at around 2-3 months after birth, reaching a plateau at around 3-9 months. During this accelerated period, there is an exponential increased synaptic density in striate cortex and frontal cortex with a parallel increased incidence of epileptic spasms during this 2-5-month period postnatally (Frost and Hrachovy, 2005). Thus, the immature cortex in the setting of brain injury, whether due to incomplete myelination or synaptogenesis, appears to be a prerequisite for the genesis of ES, as adults generally do not develop epileptic spasms after diverse causes of cerebral insult. Last, but not least, there is likely an unidentified genetic predisposition that may be a factor in the generation of epileptic spasms in the setting of the aforementioned interaction between cortical lesion, CNS maturation, and brainstem (figure 1).
Conclusion
The risk of PTE is 47/321 (14.6%) and the specific risk of ES after TBI is 6/321 (1.8%). The risk of ES appears to be high if the age at which severe TBI occurred was during infancy, and is independent of gender, type of injury (NAT vs AT), or presence/absence of skull fracture. While posttraumatic epilepsy (not ES) may start 10 years after the head injury, ES starts within two years according to our small cohort. Thus, one may anticipate peak incidence of ES after PTE between two months and two years after TBI. Epileptic spasms can start in a previously developmentally normal child even after a TBI at the age of 8 years. The pathophysiology of ES is unknown, however, our data support a combination of previously proposed models in which the primary dysfunction is a focal or diffuse cortical abnormality, coupled with its abnormal interaction with the subcortical structures and brainstem at a critical maturation stage. Furthermore, the specific maturation stage at the time of ES onset parallels a progressive, regional and lobar myelination/synaptogenesis pattern, which continues beyond toddler years. Accordingly, TBI in the more mature brain is less likely to result in ES.
Due to the retrospective nature of our study at a single level IV paediatric referral epilepsy centre, our data may not be applicable to all paediatric patients with PTE and ES across the country. Based on this small sample, the demographic and clinical characteristics may be useful to identify those at higher risk of developing ES. Patients with PTE and ES are a unique group that needs further investigation involving multiple centres.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Disclosures
None of the authors has any conflict of interest to disclose.