Epileptic Disorders
MENUDe novo 8p23.1 deletion in a patient with absence epilepsy Volume 19, numéro 2, June 2017

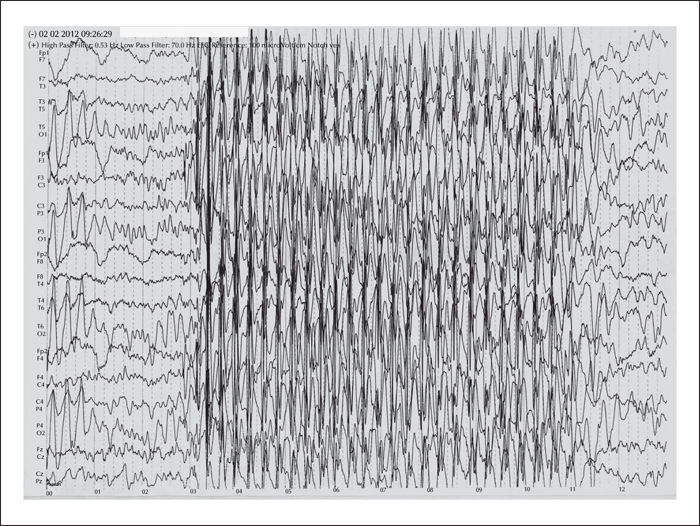
Typical absence seizures are a sudden, brief impairment of consciousness, lasting only 3-25 seconds, without a concomitant postictal phase, and occur multiple times per day. Seizures are aggravated by hyperventilation, and EEG shows a typical generalized, synchronous, bilateral 3-4 Hz spike and slow-wave discharges. Absence seizures are a type of idiopathic generalized epilepsy and are mostly seen in childhood absence epilepsy (CAE). CAE is the most common childhood epilepsy that generally begins between 3 and 9 years of age (Loiseau et al., 1995; Shinnar et al., 2015). Children with CAE have a normal neurological examination and normal intelligence level, however, frequent seizures may affect mental performance (Loiseau et al., 1995; Glauser and Loddenkemper, 2013). Cognitive changes, as well as linguistic and behavioural problems, may rarely be seen in patients with CAE (Shinnar et al., 2015).
The idiopathic generalized epilepsies have a predominantly genetic aetiology (Hughes, 2009). Mutations and susceptibility alleles have been identified in calcium channel, chloride, and GABAA receptor genes, and the functional analysis of the variants of these ion channels has revealed increased excitatory activity in the cortex or thalamocortical regions (Maljevic et al., 2006; Urak et al., 2006; Hughes, 2009). To date, GABRG2, GABRA1, GABRB3, CLCN2, and CACNA1H genes and the 8q24 region have been reported as susceptibility loci in the development of CAE (Sugimoto et al., 2000; Maljevic et al., 2006; Urak et al., 2006; Hughes, 2009). Genomic copy number variations (CNVs) constitute an important genetic risk factor for common genetic generalised epilepsy syndromes (Lal et al., 2015) and absence epilepsies (Addis et al., 2016). The recurrent hotspot microdeletions disrupt genes associated with neuronal development and function, suggesting fundamental neurodevelopmental pathways that may be important in epileptogenesis of absence seizures.
The 8p23.1 microdeletion is a rare syndrome with the phenotypic characteristics of congenital heart disease (CHD), intellectual disability (ID), behavioural problems, and microcephaly due to loss of multiple neighbouring genes (Ballarati et al., 2011). Microcephaly, physical dysmorphic anomalies, CHD, and behavioural problems may be mild or absent in this syndrome (Ballarati et al., 2011). The severity of ID and the occurrence of microcephaly appear to be related to the extent of the deletion (Claeys et al., 1997). To the best of our knowledge, there are two reports of 8p23.1 microdeletion patients in the literature; one on a patient with EEG anomalies and another on two patients with absence epilepsy (Fagan et al., 1988; Claeys et al., 1997), however, a critical region or gene that causes epilepsy has not been identified yet. Besides having typical absence seizures and EEG anomalies, our patient also suffered from serious behavioural problems, microcephaly, and minor dysmorphic features, which is why we decided to pursue genetic analysis.
Case study
A 7-year-old male patient, who was admitted to the Department of Child Neurology, Istanbul Faculty of Medicine, presented with sudden impairment of consciousness, as well as five-second periods of unresponsiveness, occurring at least 10 to 30 times per day for about six months. The antenatal and natal history was uneventful. He had undergone surgery for an ostium primum atrial septal defect, mitral valve cleft, and pulmonary artery stenosis when he was 6 months old. The patient had mental and motor developmental delay. He had mild ID and behavioural problems such as hyperactivity, outbursts of aggressiveness, and impulsiveness. He had a head trauma at the age of 5 due to a fall from a h8. Born to non-consanguineous parents, he had two healthy sisters and a brother. There was no family history of neurological or hereditary diseases or febrile convulsions.
The patient's general physical examination revealed microcephalia, high-arched palate, small ears, and swan neck deformity of the fingers. His neurological examination was normal. His IQ score was 65. Standard EEGs showed bilateral temporal-parietal-occipital and centro-temporal unconnected multiple epileptogenic foci and generalized 3-Hz spike and slow-wave discharges (figure 1). There were no structural lesions except for a post-traumatic, right frontal cortico-subcortical gliotic lesion on brain MRI which was approximately 1 mm wide. During 11 years of follow-up, he was treated with valproic acid and did not develop any other types of seizure. The seizures were almost fully controlled during this period of time.
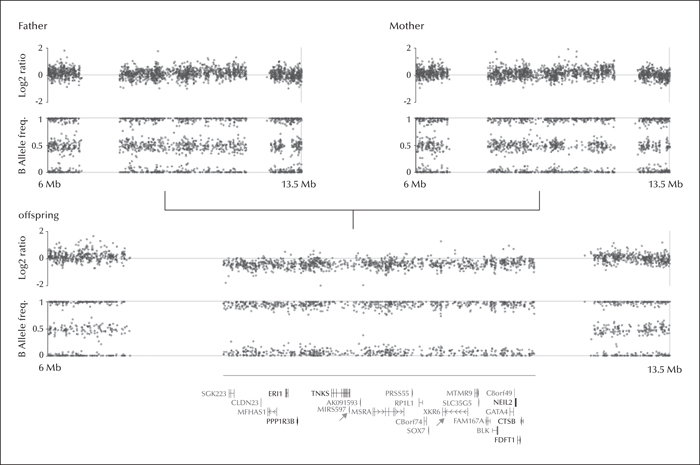
After obtaining written informed consent, the patient, mother, and father all underwent genotyping based on Affymetrix AXIOM genotyping array. The Axiom CNV summary tools (Affymetrix, Santa Clara, CA, USA) were used for calculating log2 ratios of signal intensities and B allele frequencies obtained from genotyping data. CNV calling was carried out using the Nexus Copy Number package version 7.5 (BioDiscovery Inc., Hawthorne, CA, USA). CNV screening revealed a 4-Mb de novo deletion of the chromosomal region 8p23.1 (8,109,255-12,166,733 bp [GRCh37/hg19]) in the offspring (figure 2). There were no other CNVs observed in the patient and the screening of exons of the GABRAG2 gene by PCR and Sanger sequencing gave negative results.
Discussion
Careful clinical investigation suggested that the index patient was affected by a multisystem disorder consistent with the 8p23.1 microdeletion syndrome. The 8p23.1 microdeletion is a contiguous gene syndrome which shows multiple and unrelated clinical features due to loss of multiple neighbouring genes (Ballarati et al., 2011). Each of the genes gives rise to a distinct phenotype when deleted. In our patient, the deleted 4-Mb region included about 100 genes, most of which encode for non-coding RNAs. Microcephaly, physical dysmorphic anomalies, CHD, and behavioral problems may be mild or absent in this syndrome (Ballarati et al., 2011). The severity of ID and the occurrence of microcephaly appear to relate to the extent of the deletion (Claeys et al., 1997). The GATA4 and SOX7 genes are expressed in the heart and appear to function in the same pathway, contributing to heart anomalies (Claeys et al., 1997). Regarding the other genes in this region, a specific phenotype-genotype association has not yet been described.
MicroRNA genes are small non-coding RNA molecules which are responsible for regulating gene expression. The consequence of unbalanced expression of calcium channel, chloride, and GABAA receptor genes can cause absence epilepsies. MIR597 (microRNA-597) targets GABRB3, which is known to be involved in epileptogenesis of absence epilepsies (Kan et al., 2012). The deletion of this microRNA gene may therefore be involved in the pathogenesis of absence seizures in our patient, as well as in other patients with the 8p23.1 microdeletion syndrome with an epilepsy phenotype.
In addition, the protein product of the XKR6 gene, which was among the deleted genes, localizes with a plasma membrane protein of unknown function, and is expressed only in the frontal lobe (Suzuki et al., 2014). This gene has not yet been associated with a disease, but its presence in this region is remarkable. The frontal cortex is particularly associated with absence seizures (Hughes, 2009). Various near-infrared spectroscopy studies have revealed increased oxygenation in the frontal areas, and EEG studies have shown activation in dorsolateral and orbital frontal areas (Hughes, 2009). The clinical changes during ictal absence seizures, including alteration of awareness and arrest of motor initiation, may reflect frontal lobe dysfunction. The absence seizures and pattern of behavioural problems observed in our patient may also reflect frontal lobe dysfunction.
This case report strengthens the evidence that epilepsy may be part of the phenotypic spectrum of 8p23.1 deletion syndrome, and/or haploinsufficiency of several genes with functional diversity. Until further experimental evidence to the contrary is provided, it is possible that the comorbidity of rare classic 8p23.1 deletion syndrome and typical absence seizures in the index case is merely coincidental. Nevertheless, it is highly recommendable to carry out CNV and/or whole-exome sequencing (WES) for GGE plus patients, since it may have important implications on clinical practice with regards to diagnostic classification, clinical management of the syndromic multisystem disorders, and, potentially, genetic counselling.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
This work was supported by grants from the European Community (FP7 Project DESIRE, grant 602531 to author TS), the German Research Foundation (DFG) (grants SA434/ 5-1 & SA434/4-2 to author TS), the National Genome Research Network (NGFN plus: EMINet, grants 01GS08120 to author TS), the Scientific and Technological Research Council of Turkey (TUBITAK) (grant 106S027 to author HC), and Bogazici University Research Fund (BAP) (grants 540 & 662 to author HC).
None of the authors have any conflict of interest to disclose.