Epileptic Disorders
MENUDe novo 8p23.1 deletion in a patient with absence epilepsy Volume 19, numéro 2, June 2017

- Mots-clés : 8p23.1 deletion syndrome, XKR6, MIR597, absence seizure, genomic copy number variation
- DOI : 10.1684/epd.2017.0906
- Page(s) : 217-21
- Année de parution : 2017
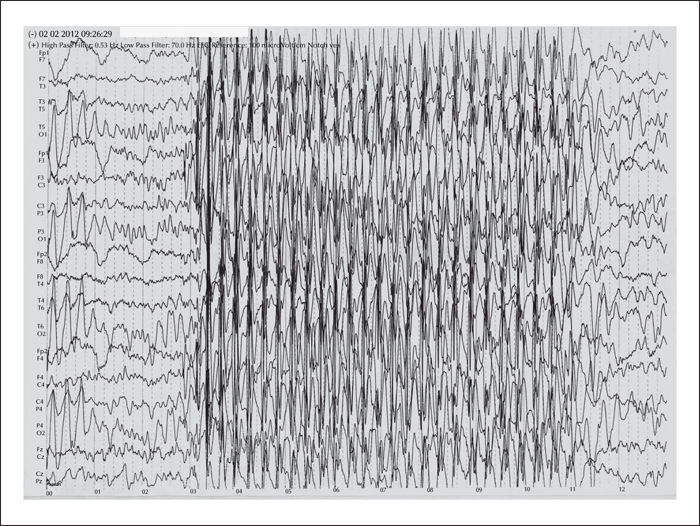
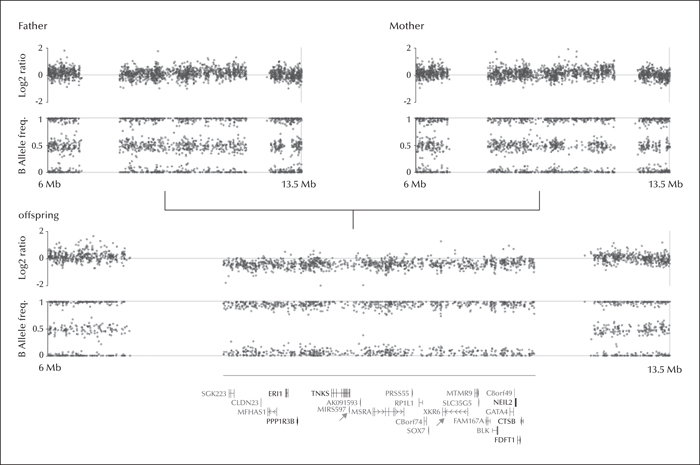
The 8p23.1 deletion syndrome is a rare multisystem disorder with high penetrance and a variable phenotypic spectrum that includes congenital heart disease (CHD), intellectual disability, behavioural problems, microcephalia, and sometimes epilepsy. Genomic copy number variations (CNVs) constitute an important genetic risk factor for common genetic generalised epilepsy syndromes (GGEs) and absence seizures. These variations, resulting either from copy loss (microdeletion) or copy gain (duplications), disrupt genes associated with neuronal development. Herein, we report an epilepsy patient who was affected by developmental delay, microcephalia, behavioural problems, CHD, and childhood-onset absence seizures. The patient had a 4-Mb de novo microdeletion at 8p23.1. Some of the genes in this region, particularly XKR6 and MIR597, may be involved in the pathogenesis of absence seizures, suggesting that epilepsy may possibly be part of the phenotypic spectrum of the syndrome rather than a comorbid disorder. Thus, CNV screening for GGE plus patients may have important implications in clinical practice with regards to diagnostic classification, clinical management of the syndromic multisystem disorders, and, potentially, genetic counselling.