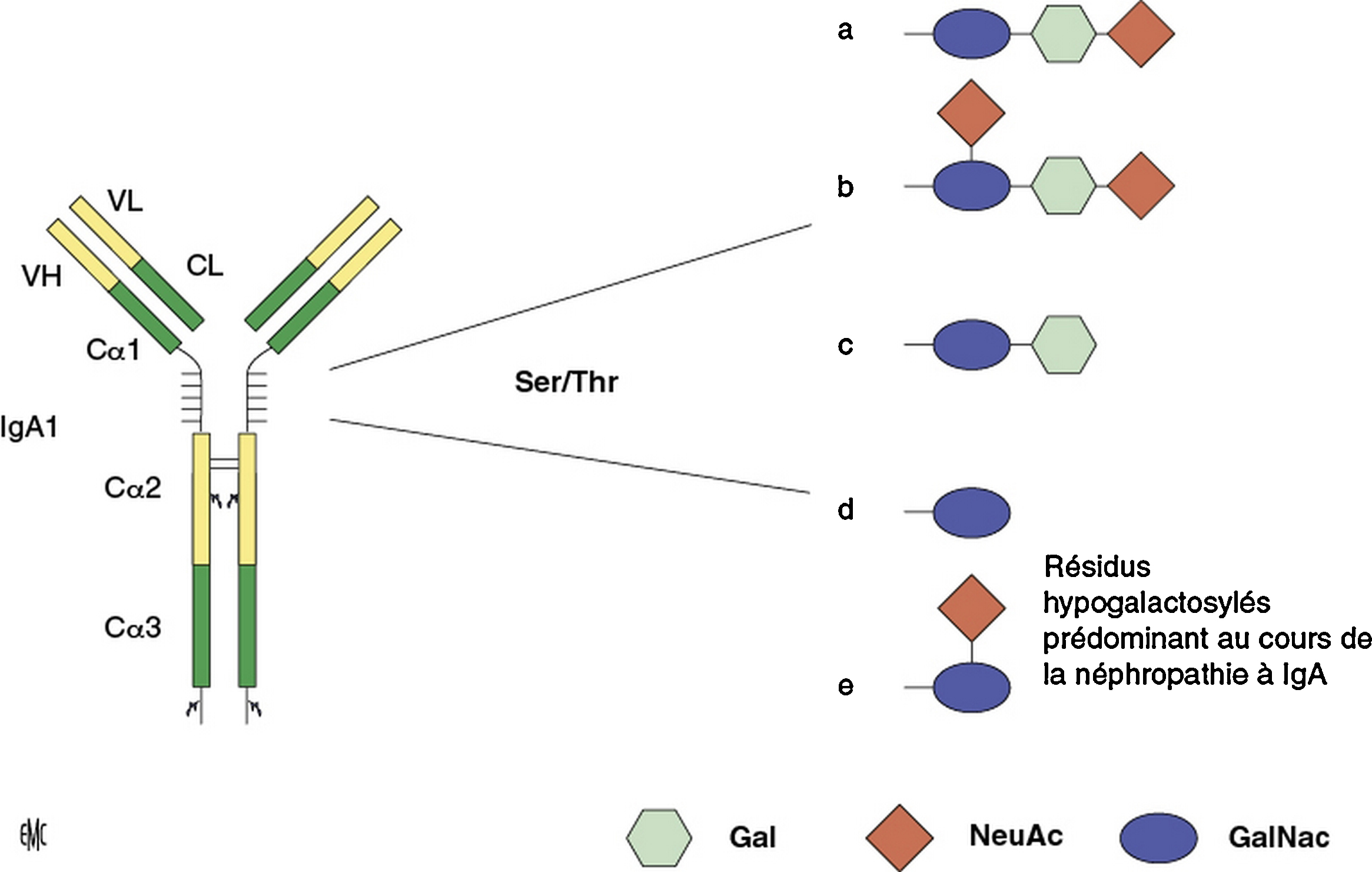
O-glycosylation de l’immunoglobuline A1 (IgA1) humaine. Les O-glycans de l’IgA1 se fixent aux résidus sérine ou thréonine de la région charnière liant les domaines CH1 et CH2 de la chaîne lourde α1 de l’IgA1. La structure de base des chaînes O-glycosylées est une N-acétylgalactosamine liant une sérine ou une thréonine. À cette structure N-acétyl-galactosamine peut s’ajouter du galactose en position β1.3 ou un acide sialique (acide N-acétylneuraminique) en position α2.6. Elle peut également être prolongée par l’acide sialique et le galactose, en position α2,3. Au cours de la néphropathie à IgA, ce sont les formes hypogalactosylées qui prédominent [47]. VH : variable heavy chain region ; VL : variable light chain region ; CL : constant light chain region ; Gal : galactose ; NeuAc : acide N-acétylneuraminique ; GalNAc : N-acétyl-galactosamine.
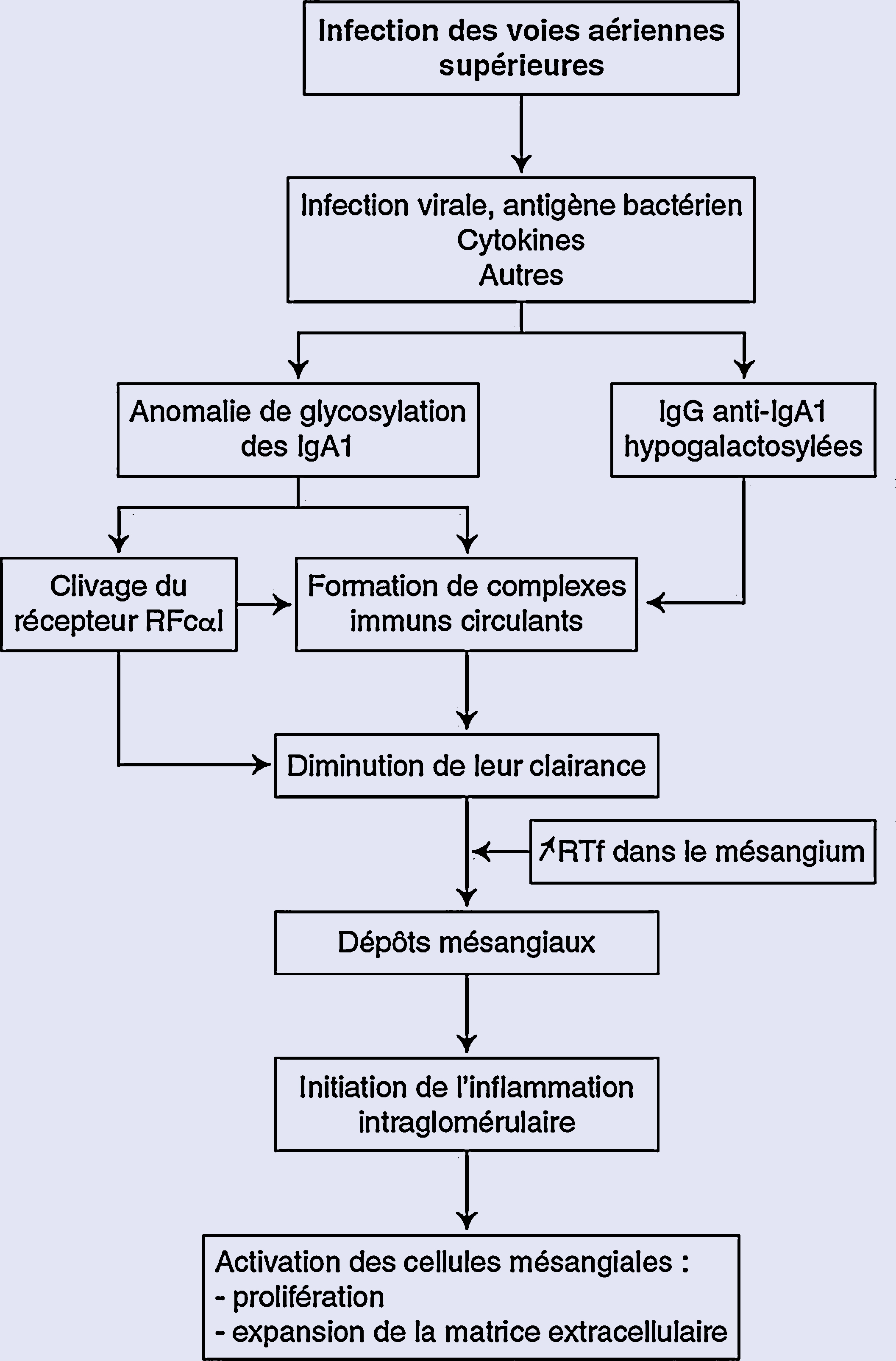
Arbre décisionnel. Physiopathologie de la néphropathie à immunoglobulines A (IgA). Un des mécanismes proposés : le rôle des anomalies de la glycosylation des IgA1. RFc : récepteur des IgA ; RTf : récepteur de la transferrine.
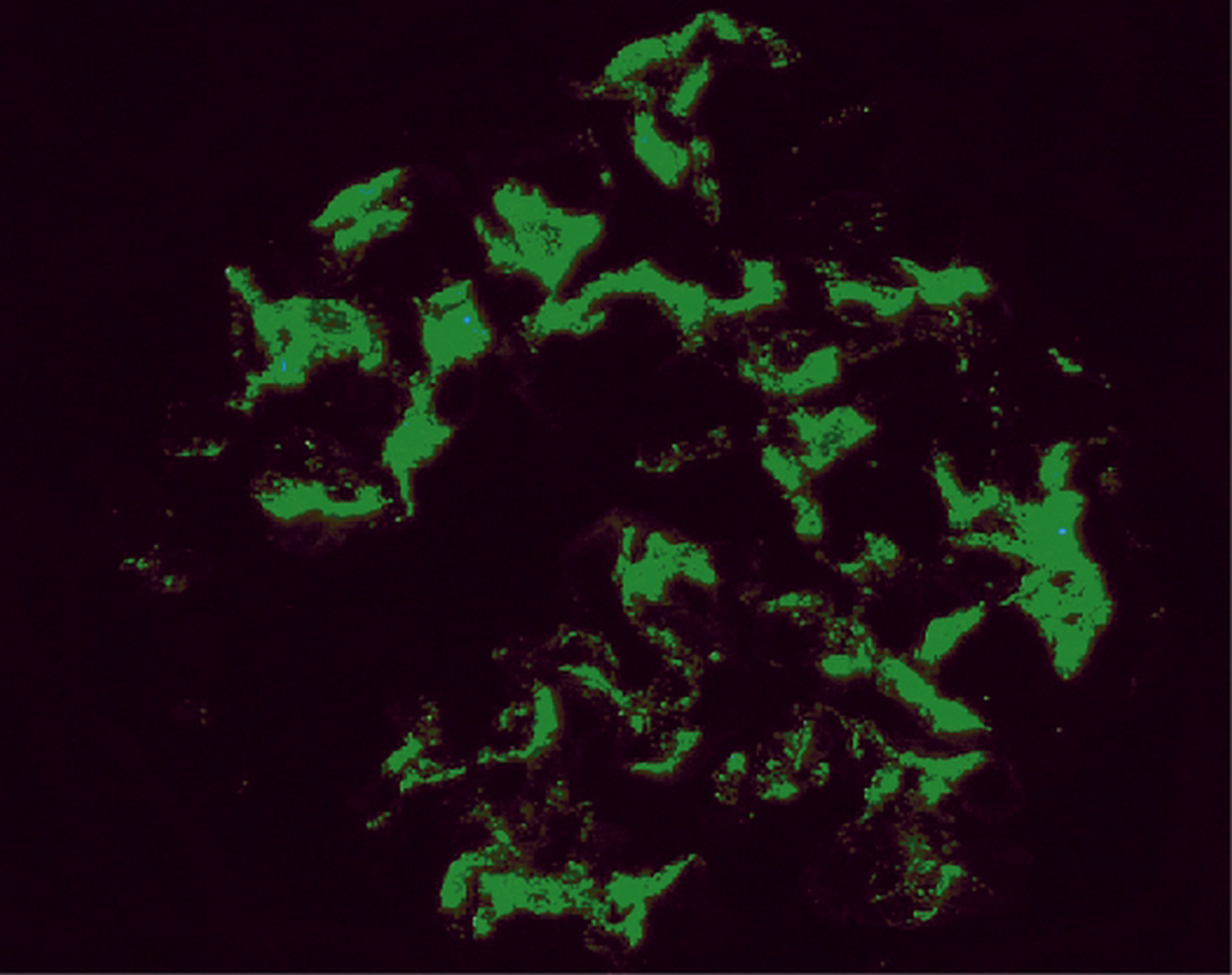
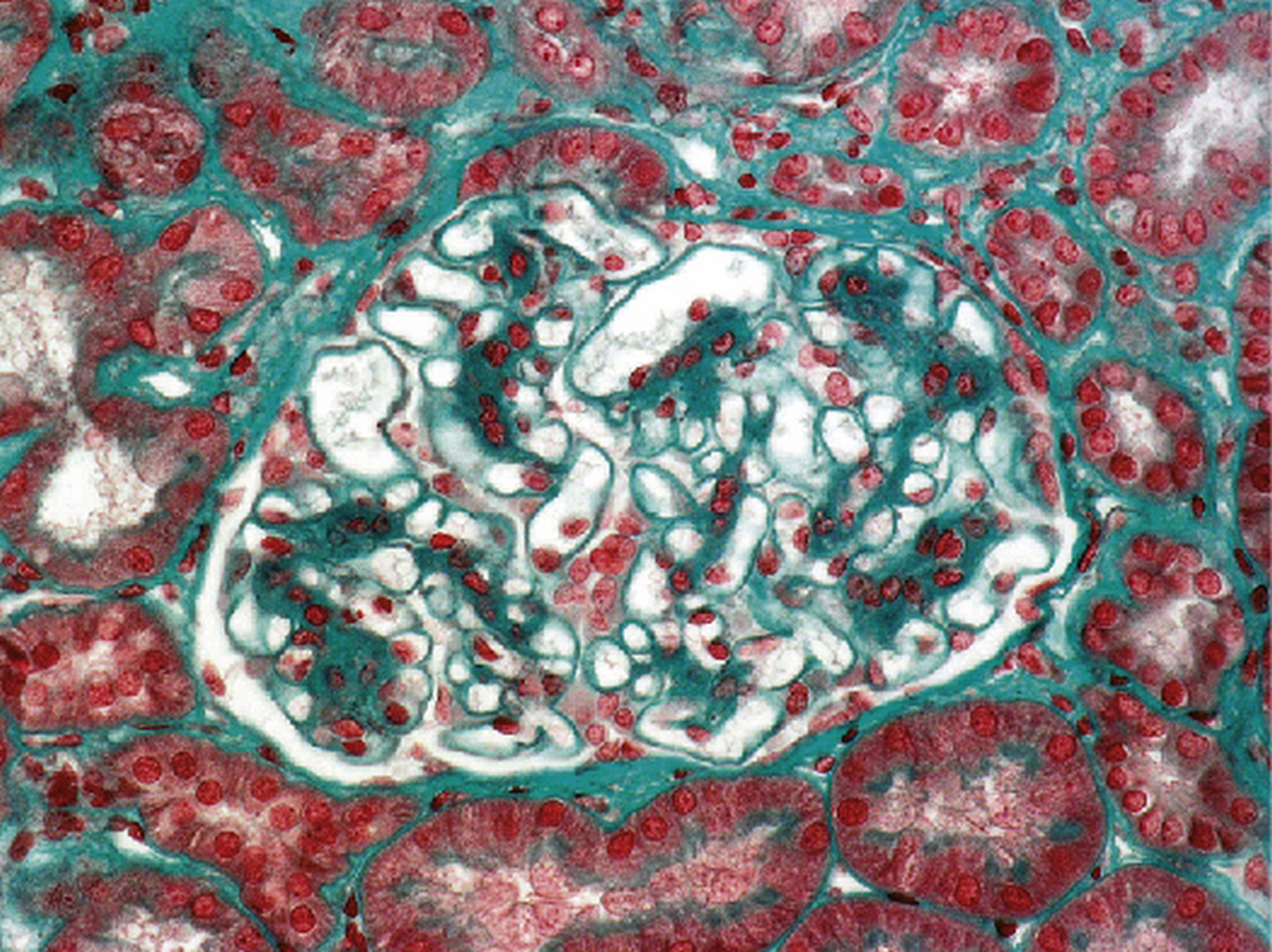
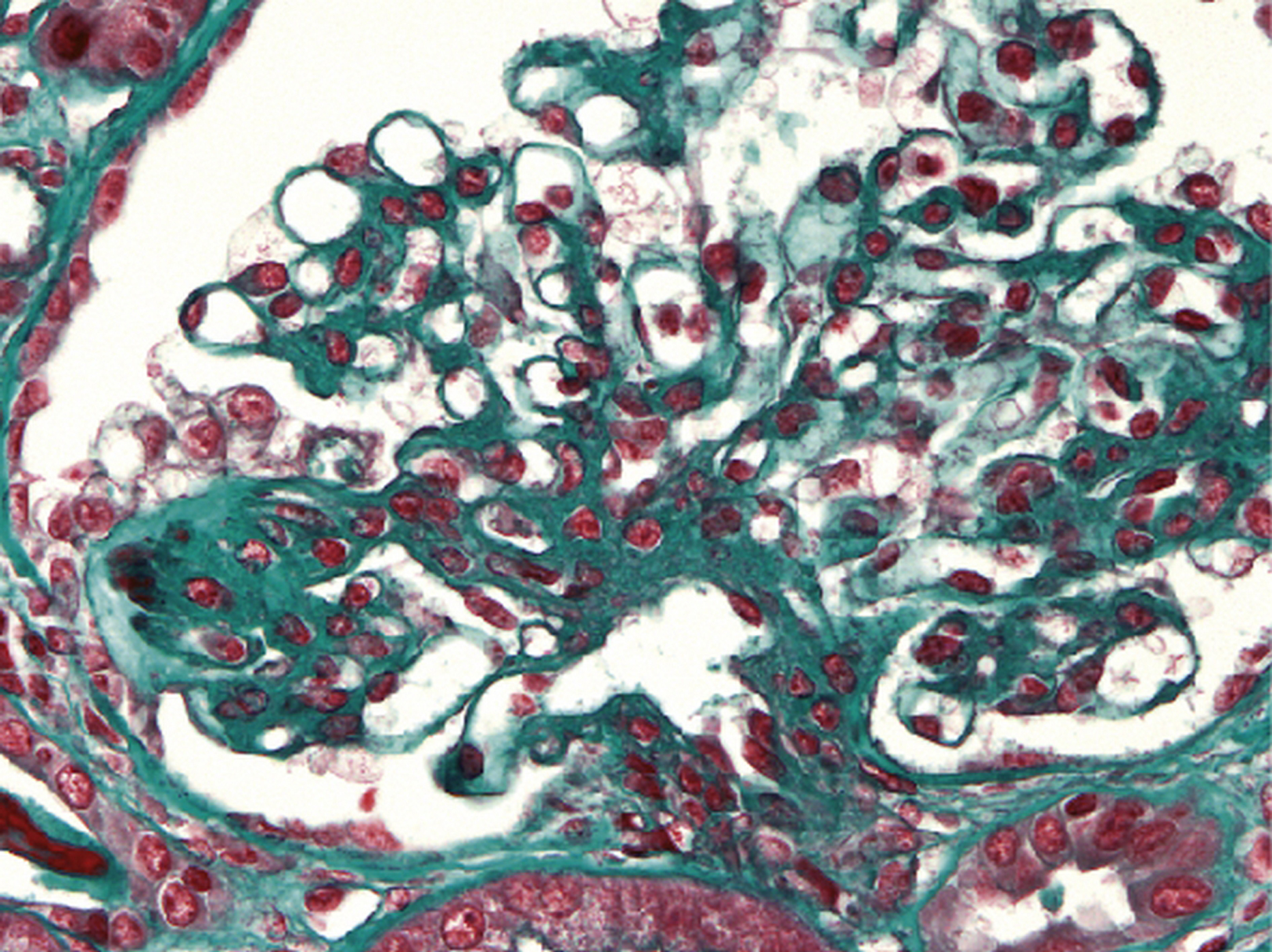
Dépôts fibrinoïdes (rouges au trichrome de Masson) de taille variable disposés dans plusieurs axes mésangiaux, correspondant à des complexes immuns contenant de l’immunoglobuline A (trichrome de Masson × 1000).
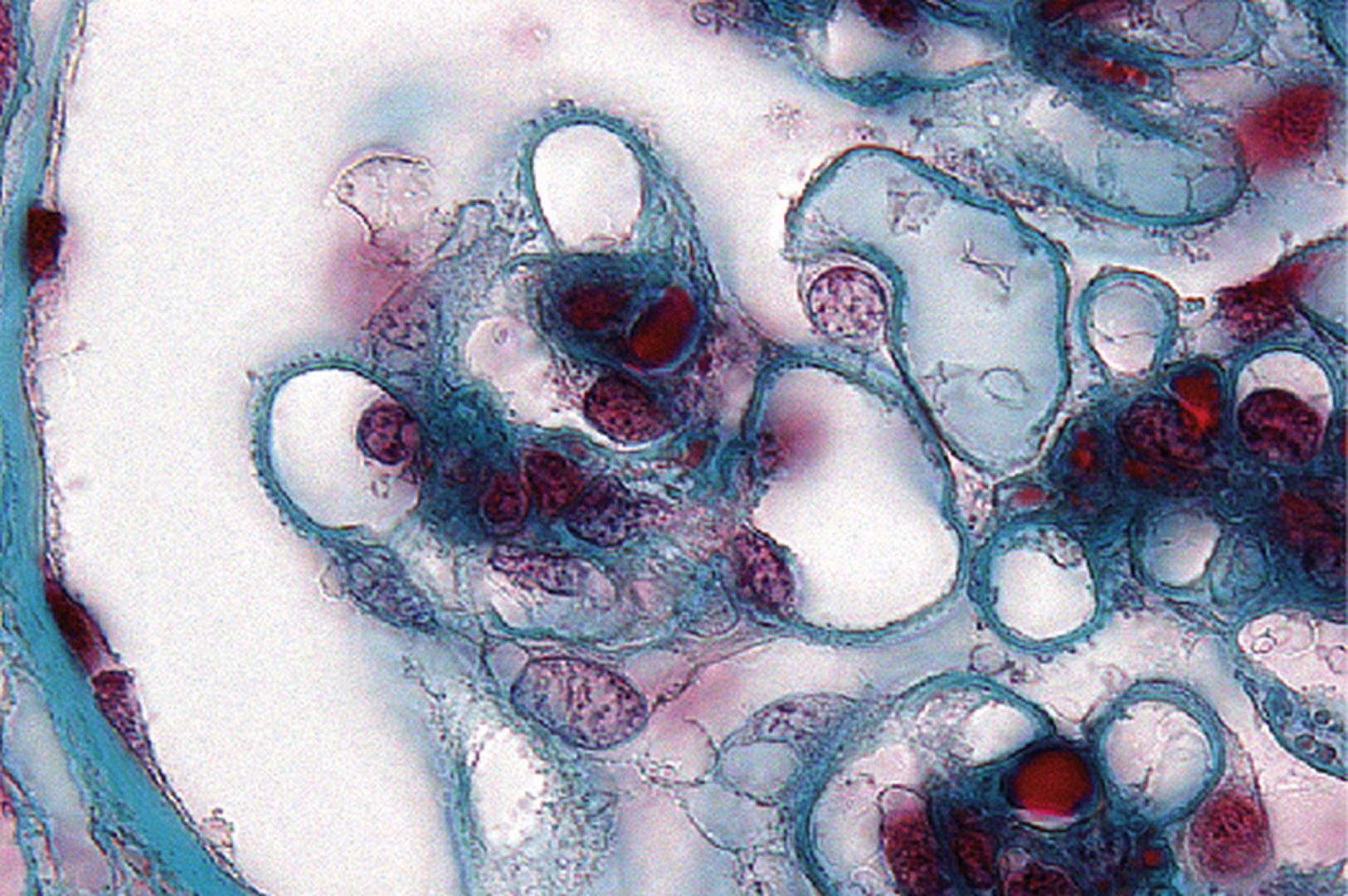
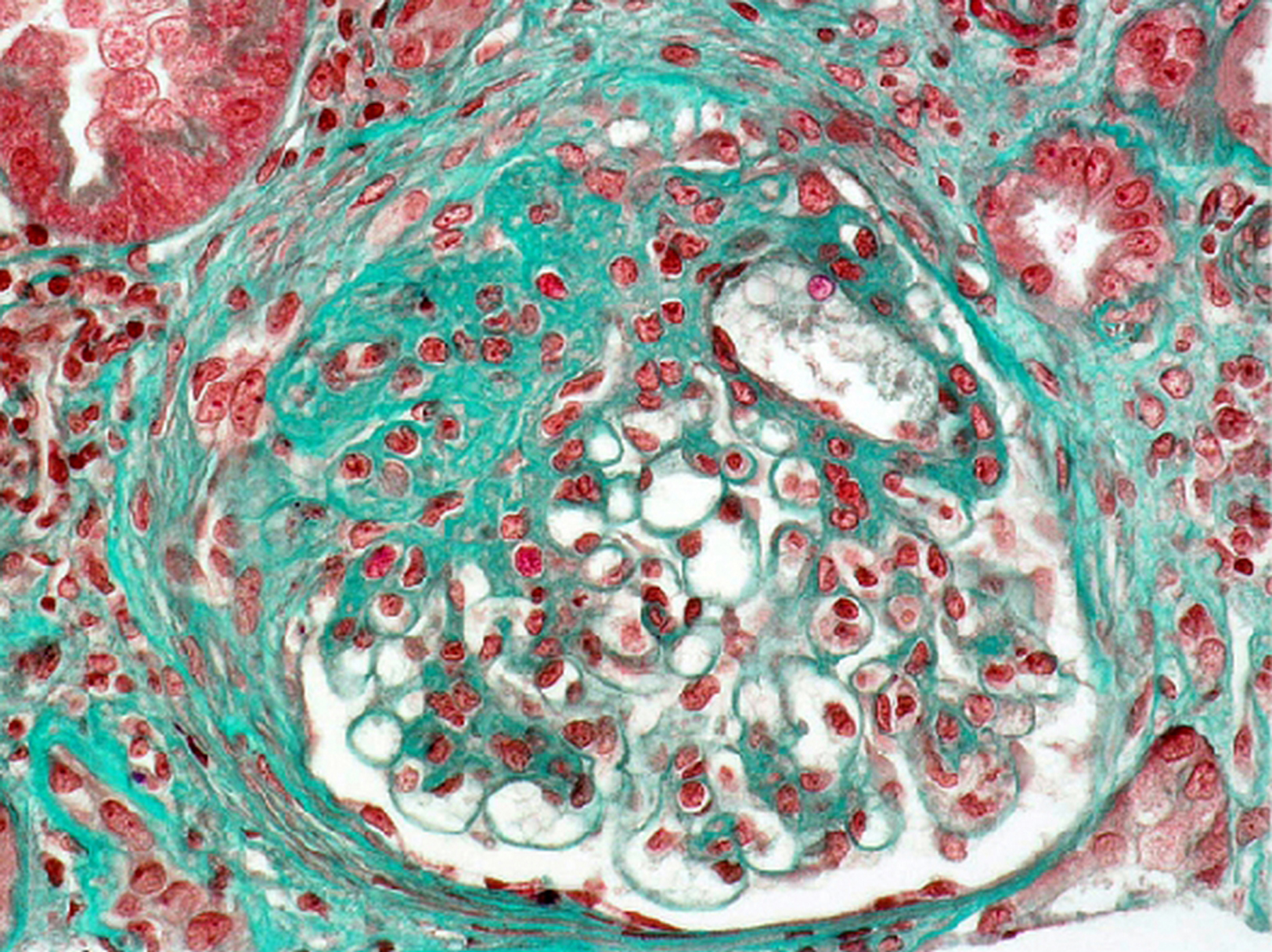
Forme mésangiale pure : un glomérule montre un épaississement net de ses aires mésangiales associé à une hypercellularité mésangiale (trichrome de Masson × 200).
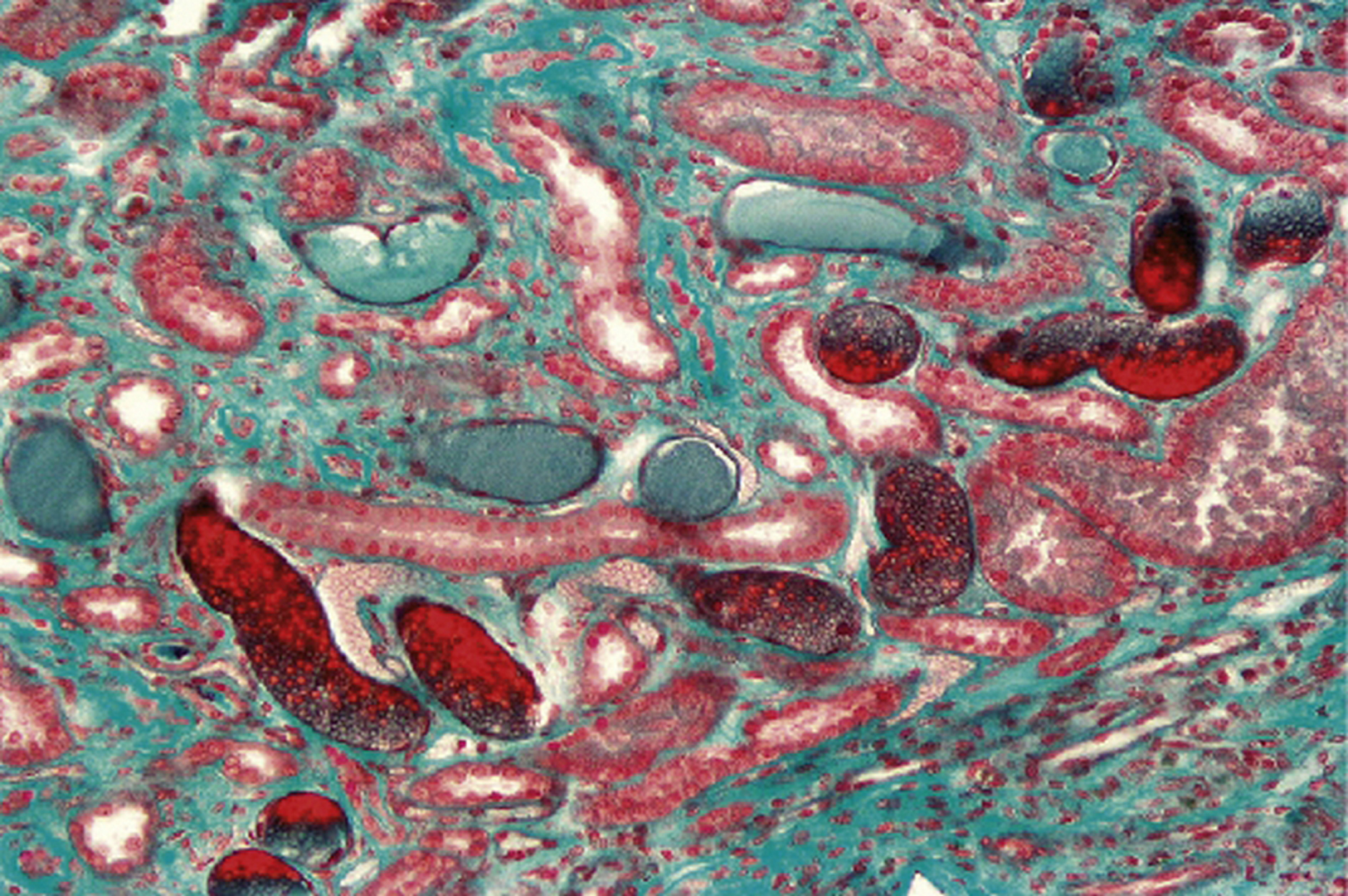
Présence de nombreux cylindres hématiques. Biopsie réalisée devant une hématurie macroscopique apparue au décours d’une angine (trichrome de Masson × 200).
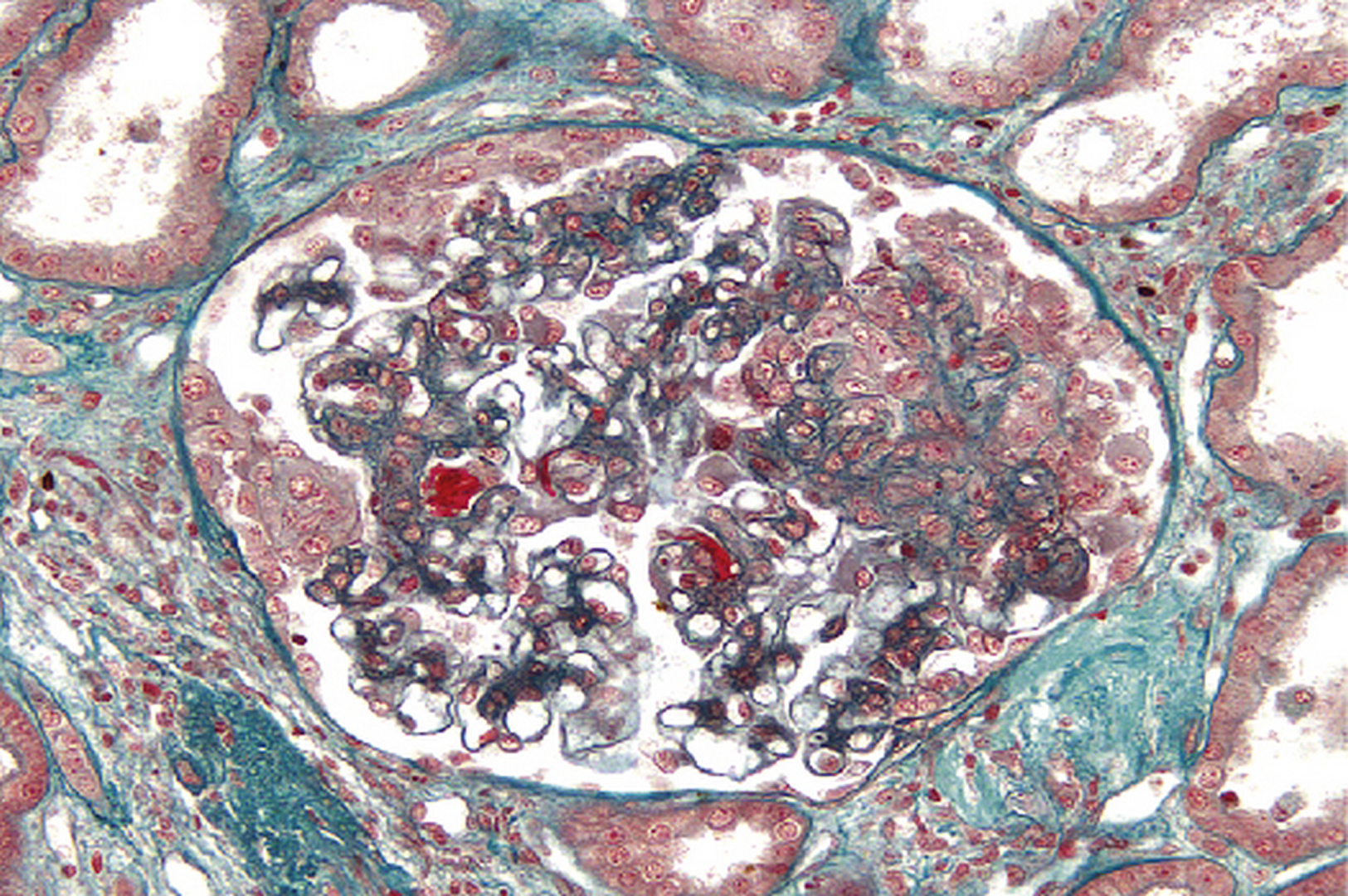
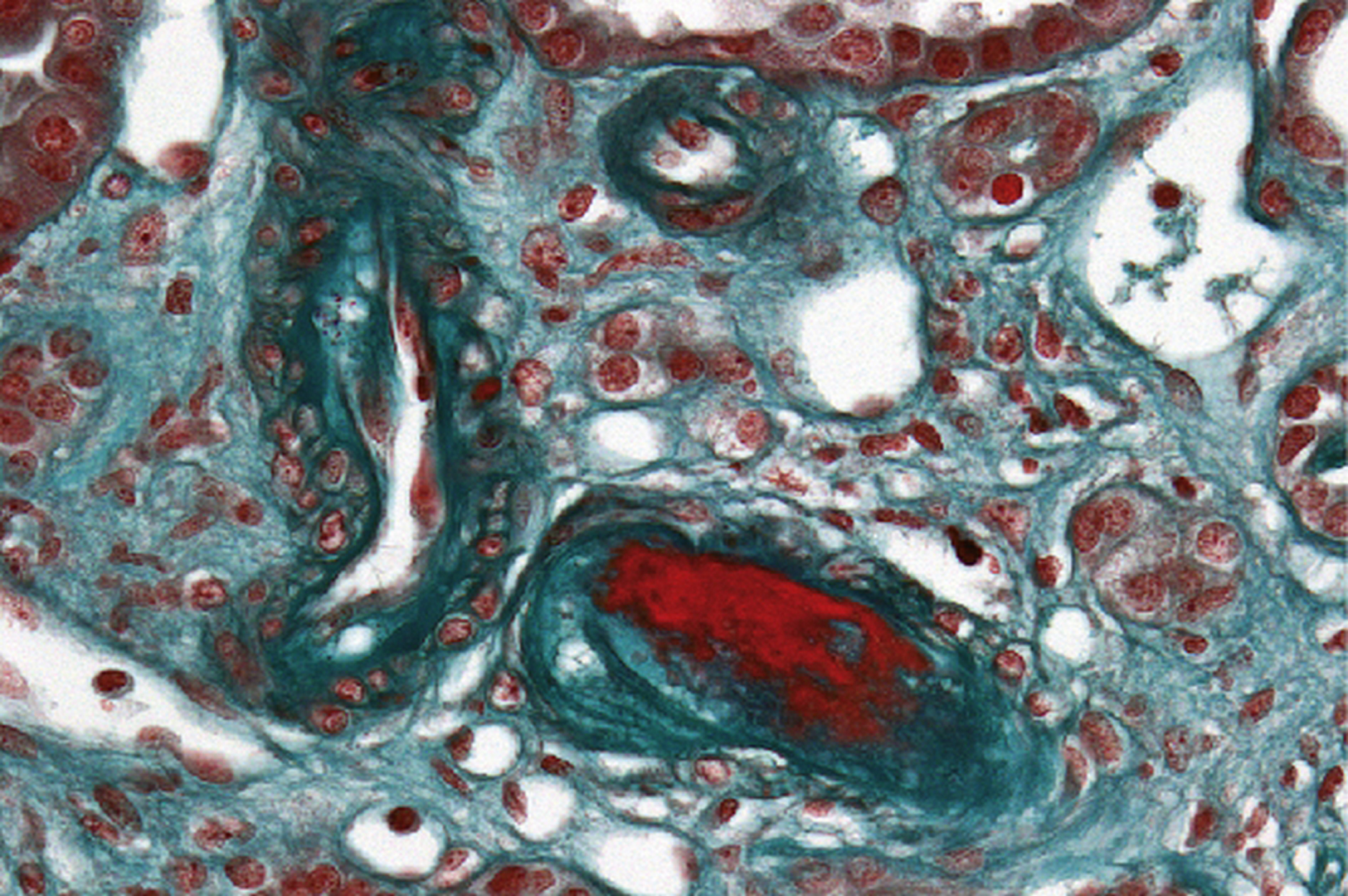
Biopsie faite au cours d’une poussée d’hypertension artérielle compliquée d’insuffisance rénale aiguë. Lésion de microangiopathie thrombotique d’une artériole juxtaglomérulaire (trichrome de Masson × 400).
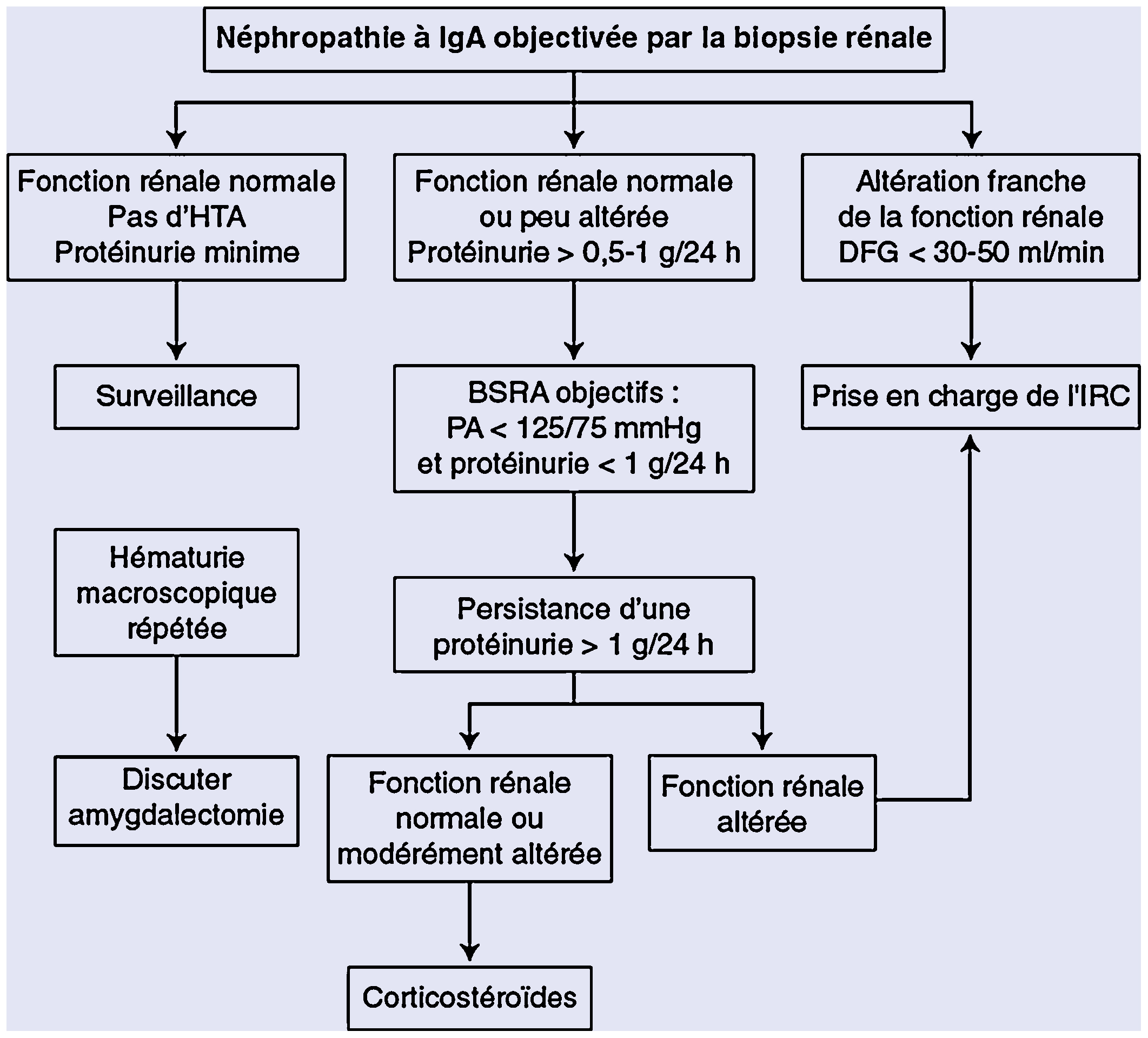
Arbre décisionnel. Prise en charge thérapeutique de la néphropathie à immunoglobulines A (IgA) [106]. BSRA : bloqueur du système rénine-angiotensine ; DFG : fonction rénale estimée par la clairance de créatinine ; HTA : hypertension artérielle ; IRC : insuffisance rénale chronique ; PA : pression artérielle.
Néoplasies : carcinome pulmonaire, du larynx, du foie, du rein et du pancréas, mycosis fungoïde, myélome multiple, POEMS
Infections : VIH, lèpre, HTLV1
Maladies de système : polyarthrite rhumatoïde, cryoglobulinémie, rhumatisme psoriasique, spondylarthrite ankylosante, vascularites à ANCA ; plus rarement : syndrome de Sjögren, maladie de Behçet, syndrome de Reiter, purpura thrombopénique idiopathique
POEMS : polyneuropathie, organomagaly, endocrinopathy, monoclonal protein, skin changes ; VIH : virus de l’immunodéficience humaine ; HTLV : human T-lymphotrophic virus ; ANCA : antineutrophil cytoplasmatic antibodies.
Tableau 1
Maladies associées à des dépôts mésangiaux d’immunoglobulines A (IgA) [3].
ÂgeAncienneté des symptômesDébit de protéinurieHypertension artérielleDysfonction rénaleIndice de masse corporelle élevé
Microscopie optiqueAdhésion capsulaire et croissantsSclérose glomérulaireAtrophie tubulaireFibrose interstitielleAmincissement de la paroi vasculaireImmunofluorescenceDépôts d’IgA dans les anses capillairesMicroscope électroniqueMésangiolyseAnomalies de la membrane basale (?)
Facteurs de bon pronostic
Hématurie macroscopique répétée
Glomérules optiquement normaux
Facteurs n’ayant pas d’impact
SexeEthnieConcentration sérique d’IgA
Intensité des dépôts d’IgAAssociation à des dépôts d’IgG, IgM, C3
Ig : immunoglobulines.
Tableau 5
Facteurs pronostiques de la néphropathie à immunoglobulines A présents au moment du diagnostic [65].
Survie rénale à 10 ans, en fonction de la présence d’une dysfonction rénale (créatinine supérieure à 1,5 mg/dL), d’une hypertension artérielle ou d’un fort débit de protéinurie, dans six différentes séries publiées.
La néphropathie à immunoglobulines A (IgA) est la glomérulonéphrite primitive la plus répandue au monde. Elle est responsable d’une insuffisance rénale progressive évoluant vers l’insuffisance rénale terminale dans près d’un tiers des cas. Les mécanismes physiopathologiques de cette pathologie médiée par des complexes immuns restent encore non élucidés. La présentation et l’évolution clinique, ainsi que l’aspect en microscopie optique de la biopsie rénale peuvent être extrêmement variables, rendant toute classification histologique difficile. La plupart des études thérapeutiques n’incluent donc jusqu’à présent les patients que sur des critères cliniques de gravité. La nouvelle classification d’Oxford devrait changer cette situation. Seule la prise en charge des patients ayant une néphropathie avec lésions glomérulaires minimes et syndrome néphrotique, ou une glomérulonéphrite extracapillaire et une insuffisance rénale rapidement progressive, est consensuelle, avec traitement par corticoïdes seuls pour les premiers et associés à des immunosuppresseurs pour les seconds. Une corticothérapie peut être discutée chez les patients ayant une protéinurie supérieure à 1 g/j sans insuffisance rénale. Tous les patients atteints de néphropathie à IgA doivent bénéficier de la prise en charge globale d’une glomérulopathie chronique, comprenant notamment un traitement par bloqueur du système rénine-angiotensine en présence d’une hypertension artérielle ou d’une protéinurie.
IgA nephropathy is the most common form of primary glomerulonephritis worldwide and an important cause of chronic kidney disease and end-stage kidney failure. Its pathophysiology remains in part unsolved but it is recognized as an immune complex disease. Recent years have brought progress in the field through the discovery of several genetic susceptibility loci and the formulation of the multi-hit pathogenesis model. Presentation, clinical course and histology can be extremely variable, making any histological classification still difficult. Indeed, most therapeutic studies until now include patients based only on the severity of clinical criteria but the new classification of Oxford should change that. Only the management of patients with nephropathy with minimal change glomerular lesions and nephrotic syndrome, or extra-capillary glomerulonephritis and rapidly progressive renal failure, is consensual: Corticosteroids alone for the first and associated with immunosuppressive drugs for the latter. The recent Kidney Disease Improving Global Outcomes (KDIGO) consensus treatment guideline is still controversial, especially in light of the last clinical studies. Corticosteroid therapy can be discussed in patients with proteinuria greater than 1 g/day without renal failure. All IgA nephropathy patients should benefit from the global management of chronic glomerular disease, including a renin-angiotensin system blocker in the presence of hypertension or proteinuria.