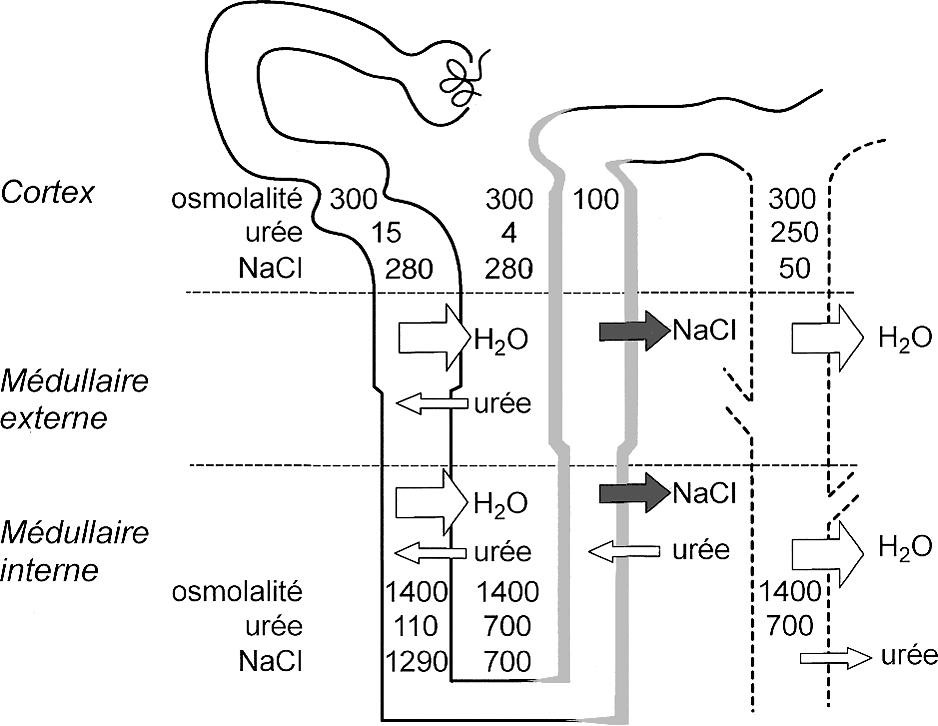
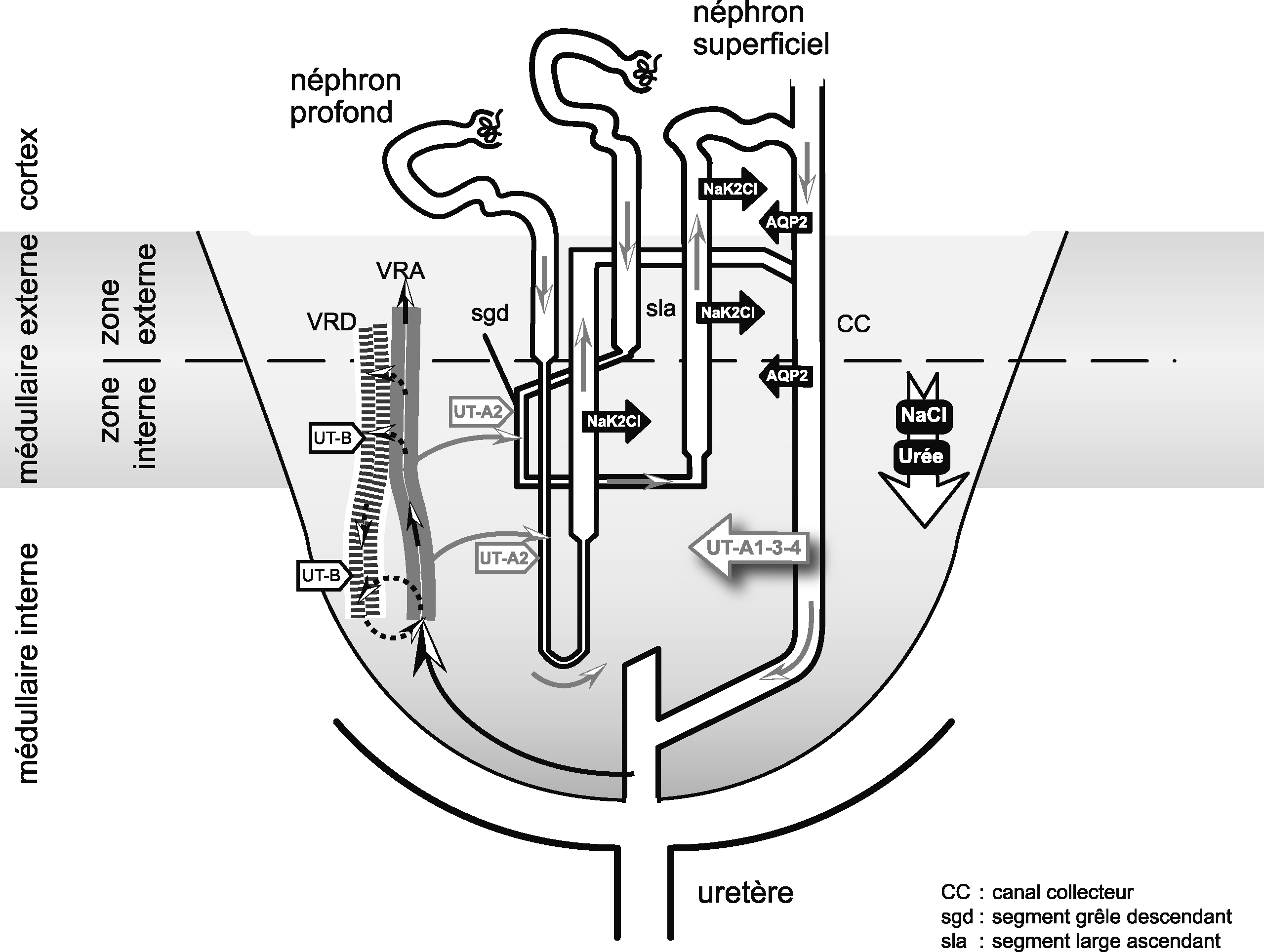
Représentation schématique d'un seul néphron et du mécanisme de concentration urinaire d'après le modèle de Kokko et Rector.
Les traits épais qui bordent la branche ascendante de l'anse de Henle indiquent une perméabilité très basse pour l'eau. Les flèches de différentes tailles indiquent les mouvements d'eau et de solutés dans les différents segments. L'osmolalité (mOsm/kg) et les concentrations en urée (mmol) et NaCl (mmol) sont représentées. Dans la médullaire externe, la branche ascendante large de l'anse de Henle réabsorbe de façon active le chlorure de sodium (Fig. 3). Il en résulte une diminution de l'osmolalité de l'urine luminale et une augmentation de l'osmolalité médullaire interstitielle. Le mécanisme passif de contre-courant proposé en 1972 par Kokko et Rector [99] et par Stephenson [100] propose la réabsorption passive de NaCl par la branche ascendante fine de l'anse de Henle car l'interstitium médullaire a une concentration plus basse en NaCl mais plus élevée en urée. Cependant, il n'y a pas eu confirmation de ce modèle de concentration passive dans les études récentes de souris invalidées à la fois pour les transporteurs d'urée UT-A1 et UT-A3 [101].
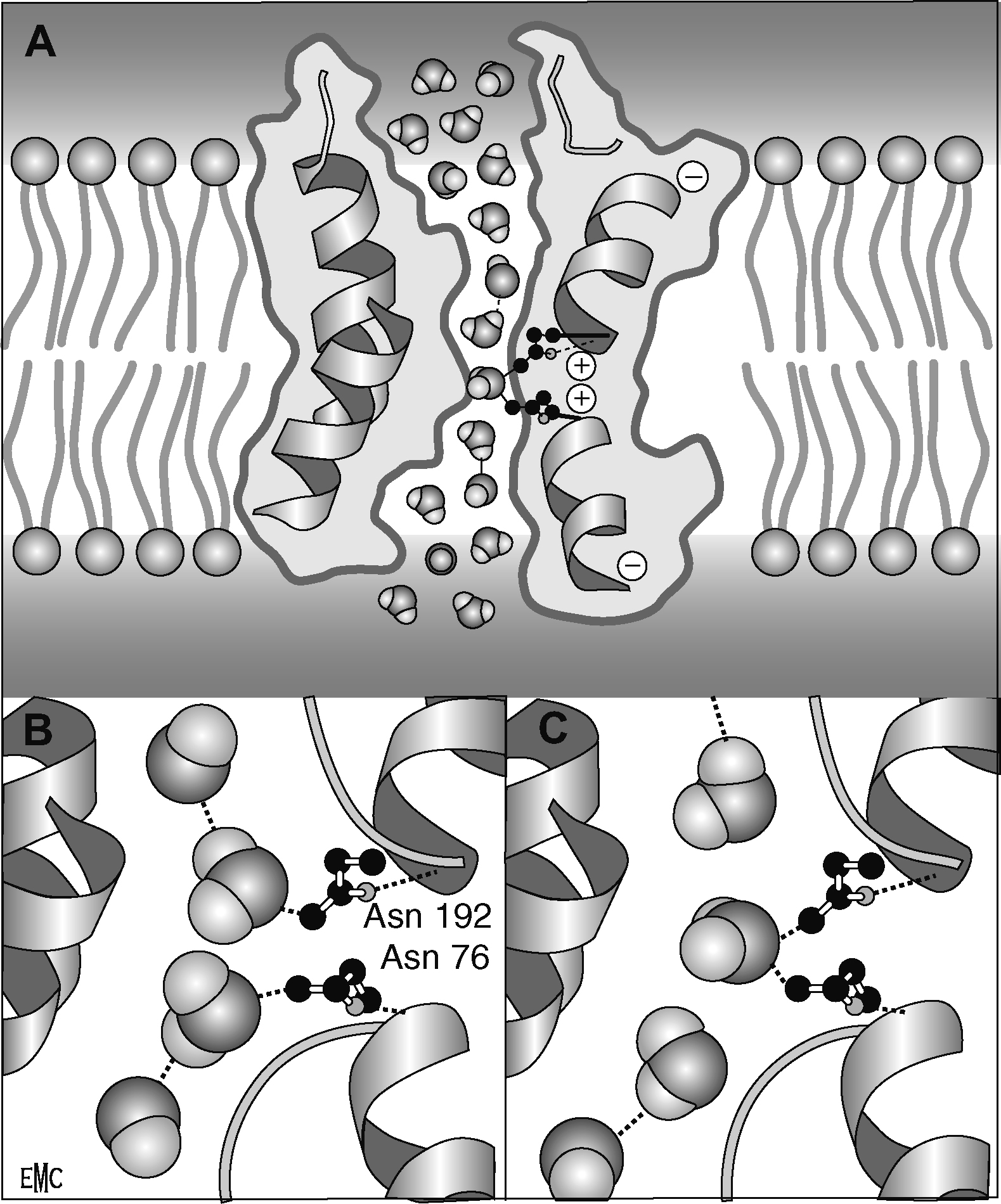
L'eau passe mais les protons ne passent pas. A. Les charges partielles au niveau des dipôles de chaque hélice réorientent les molécules d'eau qui traversent la chicane de constriction. B, C. Liaison d'une molécule d'eau aux résidus Asn76 et/ou Asn192 dont les groupements amides font protrusion dans le canal à l'eau.
Représentation préparée avec Molscript [102]. Reproduit avec permissions [103] Copyright (2000) Macmillam Magazines Ltd.
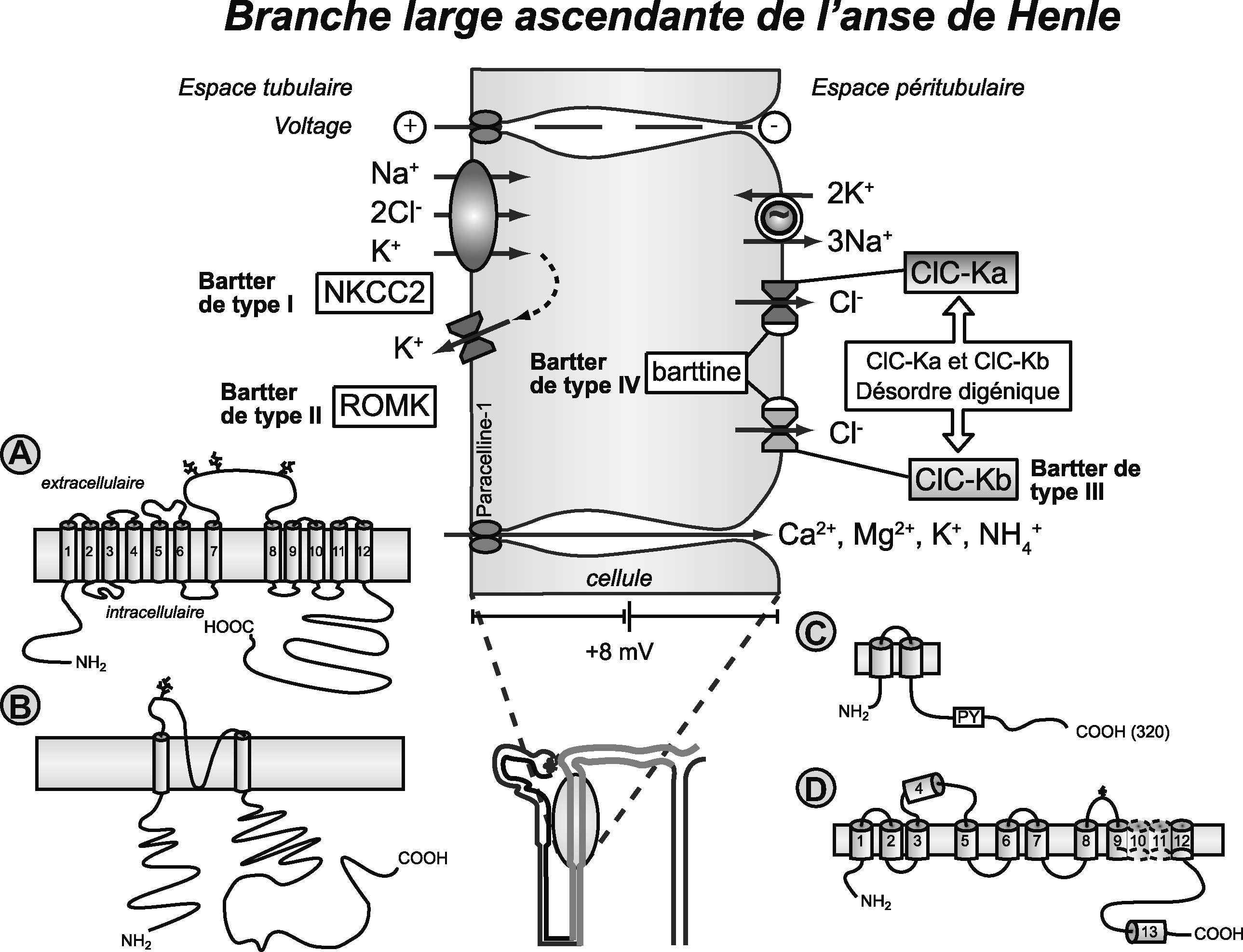
Réabsorption du sodium dans la branche large ascendante de l'anse de Henle.
Des syndromes polyuropolydipsiques d'intensité variable, parfois extrêmement sévères, sont souvent observés chez les patients avec syndrome de Bartter : la diurèse aqueuse observée s'accompagne de natriurèse et d'autres perturbations de l'équilibre hydroélectrolytique (potassium, calcium). Les syndromes de Bartter sont secondaires à des mutations avec perte de fonction de quatre gènes différents. 1) Les mutations du gène SLC12A1 qui code pour le transporteur sodium, potassium, chlorure(triporteur) NKCC2 (A), transporteur inhibé par le bumétamide ou le furosémide, sont responsables des syndromes de Bartter de type I [104]. La protéine NKCC2 est exprimée exclusivement au niveau des membranes apicales de la branche ascendante large de l'anse de Henle. 2) Les syndromes de Bartter de type II sont secondaires à des mutations avec perte de fonction du gène KCNJ1 codant pour la protéine ROMK [105,106], un canal potassique rectifiant entrant (B). Les mutations du CLCNKB codant pour le canal chlorure ClC-Kb (C) ou du gène BSND codant pour la protéine barttine (D) sont responsables respectivement des Bartters de types III [107] et IV [10,11,94,95,108] (C, D). Il existe des Bartter digénique ClC-Ka et ClC-Kb [108].
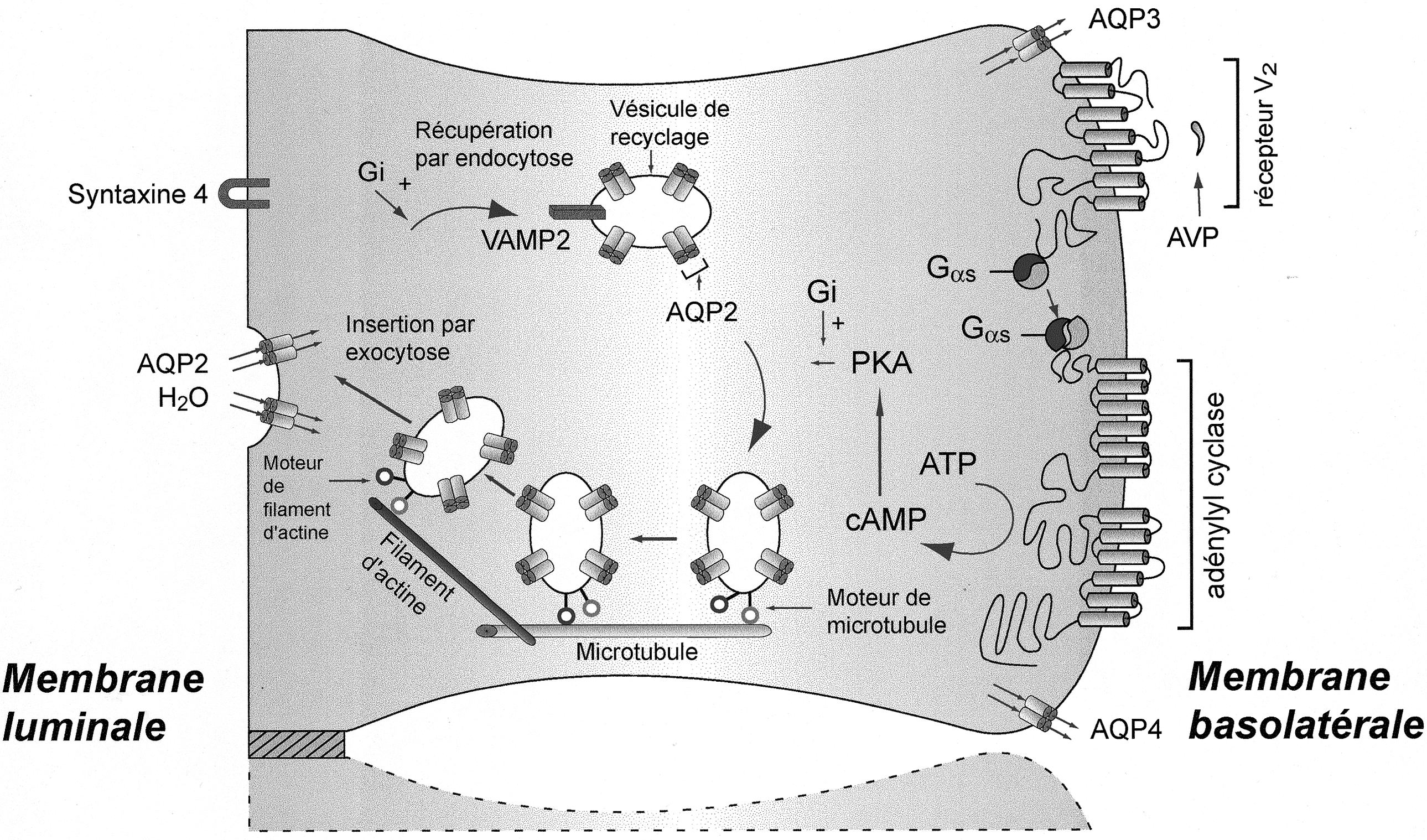
Représentation schématique d'une cellule principale du canal collecteur rénal et des éléments qui conduisent à la réabsorption transcellulaire de l'eau.
D'autres fonctions importantes de cette cellule, en particulier la réabsorption sodée par l'intermédiaire du canal épithélial sodique (ENac) et sa régulation par l'aldostérone ne sont pas représentées [109]. Le récepteur V2 à la vasopressine lie la vasopressine circulante sur le côté basolatéral de la cellule et induit la formation d'adénylate cyclase par l'intermédiaire d'une protéine G trimérique. La phosphorylation de différents effecteurs induit la fusion à la membrane de vésicule endocytaires portant les aquaporines 2. Les microtubules et les filaments d'actine facilitent le mouvement des vésicules porteuses d'aquaporine 2 à la membrane. En présence de vasopressine, la membrane luminale devient perméable à l'eau, l'eau quitte la cellule par les aquaporines 3 et 4 exprimées constitutionnellement au niveau de la membrane basolatérale. L'eau suit les caractéristiques de tonicité environnantes : la lumière tubulaire est hypotonique (beaucoup de molécules d'eau par unité de volume), l'interstitium est hypertonique (peu de molécules d'eau par unité de volume). En l'absence de vasopressine, les vésicules endocytaires contenant les AQP2 sont récupérées par endocytose dans un compartiment sous-endothélial.
Représentation schématique des transporteurs d'urée dans le néphron ainsi que les voies vasculaires et tubulaires qui contribuent au recyclage de l'urée.
Une anse de Henle courte qui atteint la limite interne de la médullaire interne, et une anse de Henle longue qui pénètre profondément dans la médullaire interne, sont représentées. UT-A1/3 et possiblement UT-A4 sont exprimés au niveau de la membrane apicale du tubule collecteur de la médullaire interne. UT-A1/3 sont responsables de la réabsorption d'urée dépendante de la vasopressine. UT-A2 est présent au niveau des branches descendantes fines des néphrons superficiels et profonds. Dans la zone interne de la médullaire externe les vasa recta descendants (VRD) et ascendants (VRA) ainsi que les segments grêles descendants (sgd) de l'anse de Henle sont proches l'un de l'autre. L'UT-B est présent dans les VRD et permet un contre-courant échangeur d'urée entre VRA et VRD ainsi qu'entre VRD et sgd. La flèche verticale sur la droite représente le gradient d'osmolalité corticopapillaire constitué principalement d'urée et de NaCl. sla : segment large ascendant ; TC : tubule collecteur. Les flèches noires, fines ; pleines indiquent la livraison et le transit d'urée dans les vasa recta ascendants à partir de la médullaire rénale. Le retour (recyclage) d'urée dans la médullaire rénale est indiqué par des flèches noires pointillées à partir des routes vasculaires ou par des flèches pleines grises à partir des routes tubulaires [110].
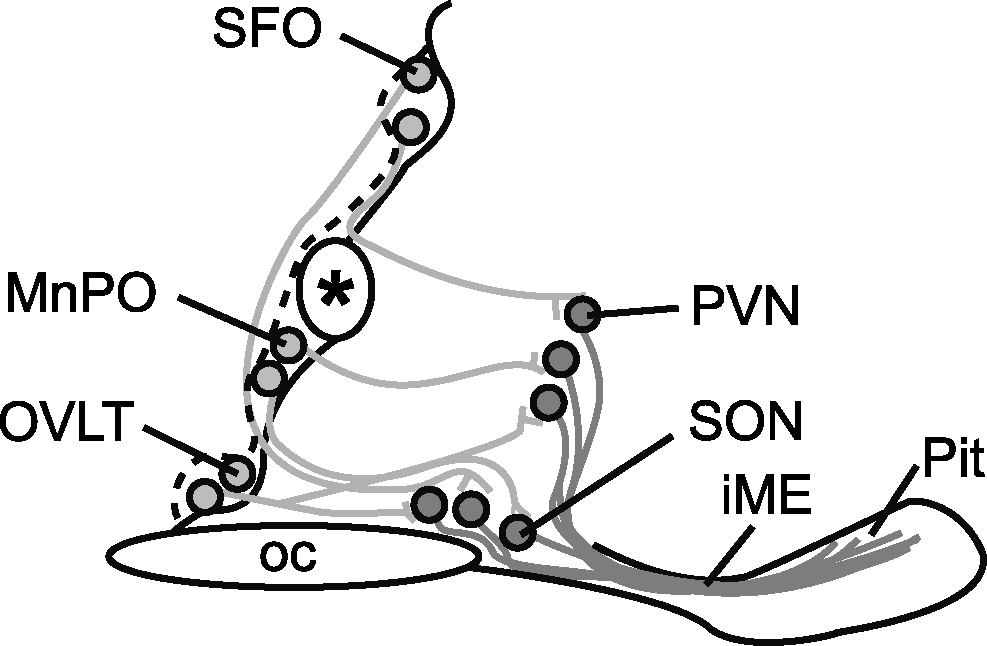
Représentation schématique des voies osmorégulatrices de l'hypothalamus, il s'agit ici d'une section sagittale du cerveau ventral au niveau du troisième ventricule chez la souris.
Les neurones de la lame terminale (lamina terminalis) — qui comprennent l'organe vasculaire de la lame terminale (OVLT : organum vasculosum lamina terminalis), le noyau préoptique médian (MnPO : median preoptic nucleus) et l'organe subfornical (SFO : subfornical organ) –– répondent à l'hypertonicité plasmatique par la voie de leurs projections axonales (lignes grises) vers les neurones magnocellulaires des noyaux paraventriculaires (PVN : paraventricular nucleus) et supraoptiques (SON : supraoptic nucleus). Les axones de ces neurones magnocellulaires forment le tractus hypoothalamoneurohypophysaire qui parcourt l'éminence médiane pour atteindre l'hypophyse postérieure, lieu de neurosécrétion de la vasopressine et de l'ocytocine [111].
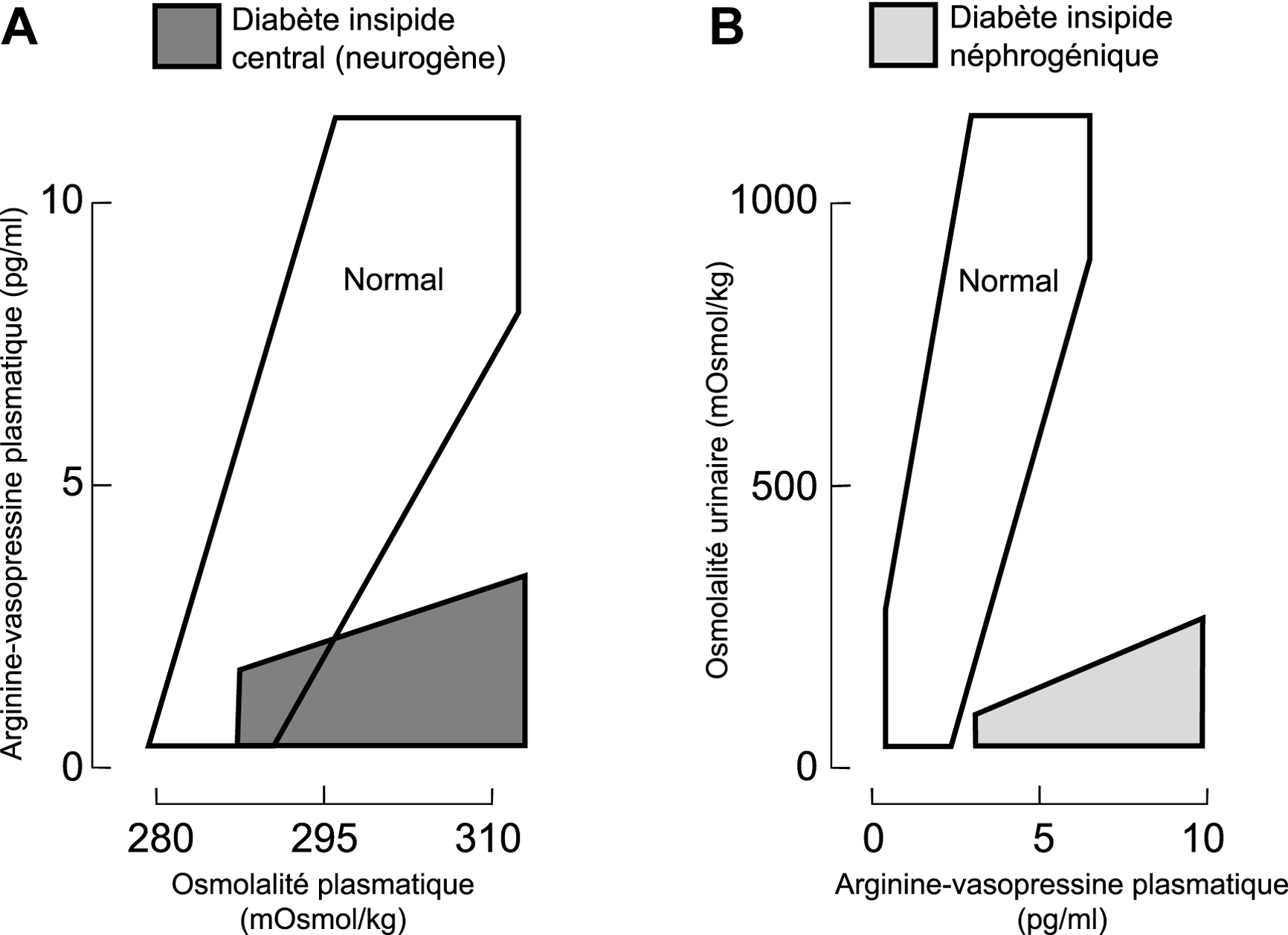
Diagnostic différentiel des polyuries : mesures de l'arginine–vasopressine plasmatique (PAVP) de l'osmolalité plasmatique (POsm) et de l'osmolalité urinaire UOsm). A. Partie gauche : corrélation entre PAVP et POsm pendant la déshydratation et/ou la perfusion de solution saline hypertonique. Les patients avec polydipsie primaire ou les patients avec diabète insipide néphrogénique (DIN) ont des valeurs normales (surface ouverte) : au contraire, les patients avec diabète insipide neurogène (central) ont des réponses subnormales (surface gris foncé). B. Partie droite : corrélation entre UOsm et PAVP pendant des épreuves de déshydratation et/ou de charge en eau. Les patients avec diabète insipide central (neurogène) et les patients avec polydipsie primaire ont des valeurs normales (surface ouverte). Au contraire, les patients avec DIN ont une urine hypotonique malgré les concentrations élevées de PAVP (surface gris clair). Figure modifiée [112].
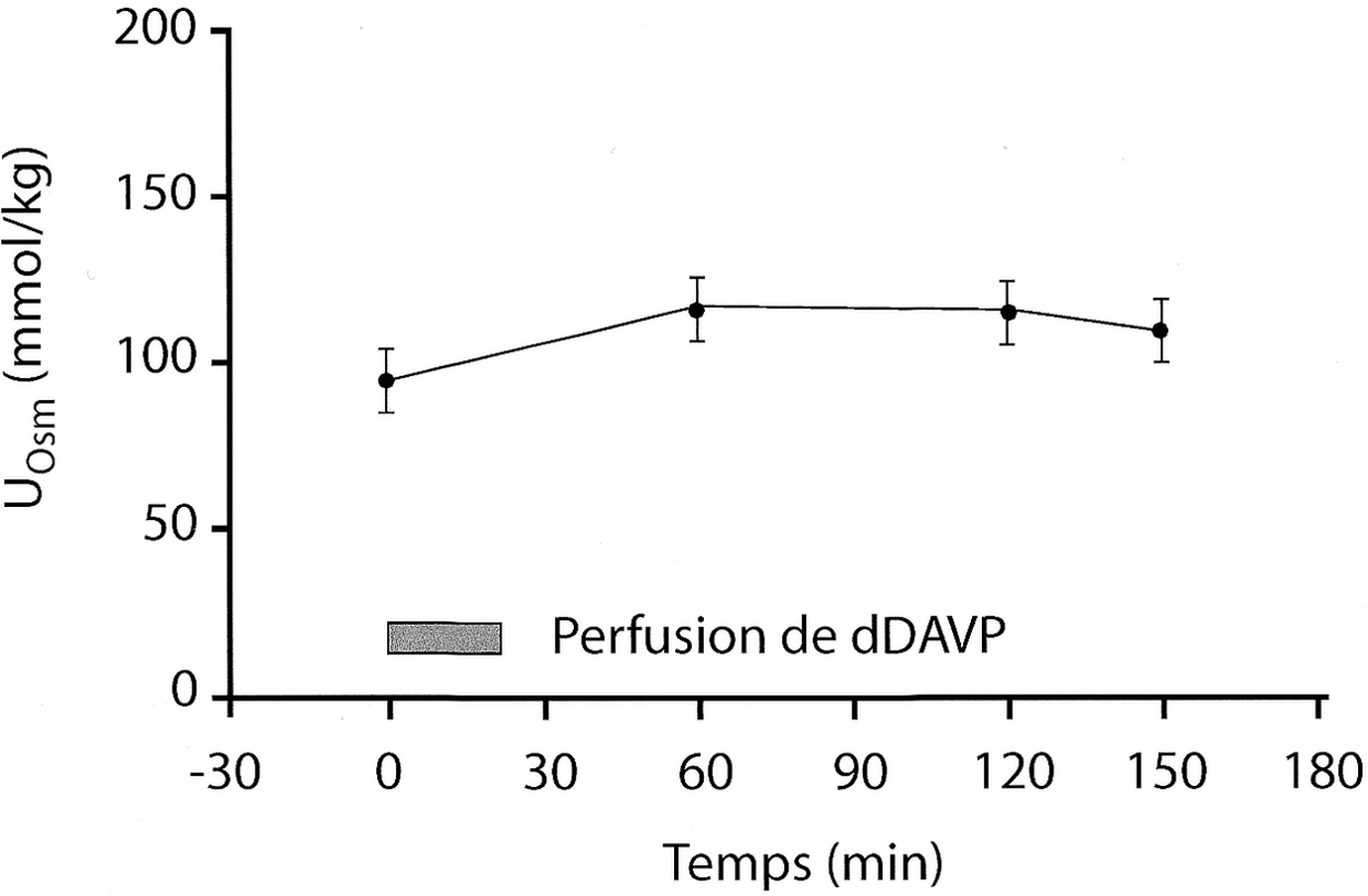
Osmolalité urinaire (UOsm) après perfusion de dDAVP (0,3 μg/kg de poids corporel jusqu'à un maximum de 24 μg perfusé de 0 à 20 minutes) chez 13 patients avec diabète insipide néphrogénique lié à l'X provenant de six familles différentes avec mutations bien identifiées du gène du récepteur V2.
L'osmolalité urinaire est inchangée de même que (non représentées) la clairance de l'eau libre et la clairance osmolaire. La moyenne et l'intervalle de confiance de la moyenne sont indiqués.
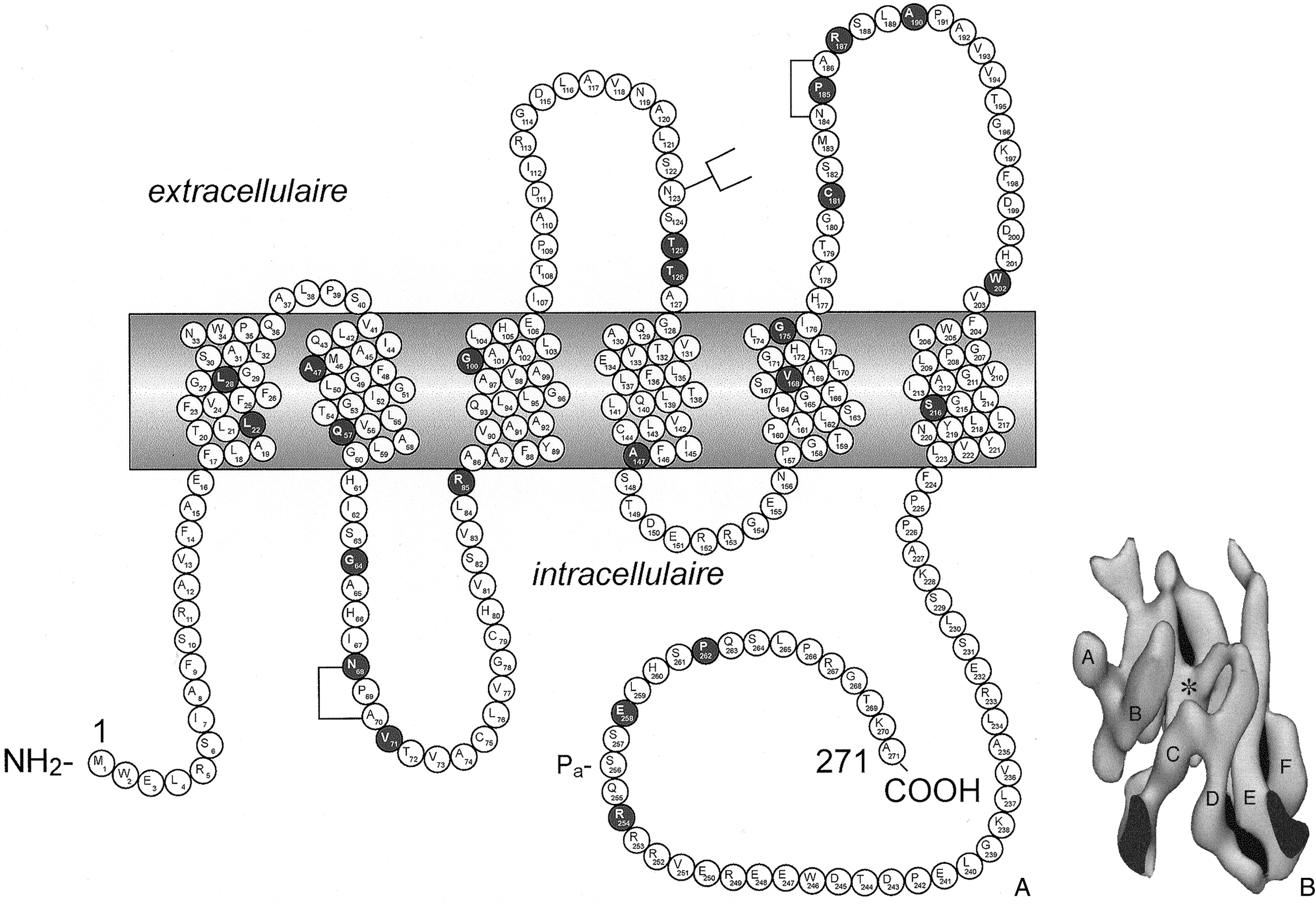
Représentation schématique plane du récepteur V2 de la vasopressine, de ses sept passages transmembranaires et de ses 371 acides aminés ; 183 mutations identifiées par des cercles gris foncé correspondant à l'acide aminé modifié (ou au début de la séquence modifiée quand il y a décalage du cadre de lecture) sont représentées.
Les acides aminés sont identifiés par un code à une seule lettre. Un chiffre sous l'acide aminé indique plus d'une mutation sur le même codon. Les mutations sont nommées d'après les recommandations publiées [113]. Les domaines extracellulaires, transmembranaires et cytoplasmiques ont été définis par Mouillac et al. [114].
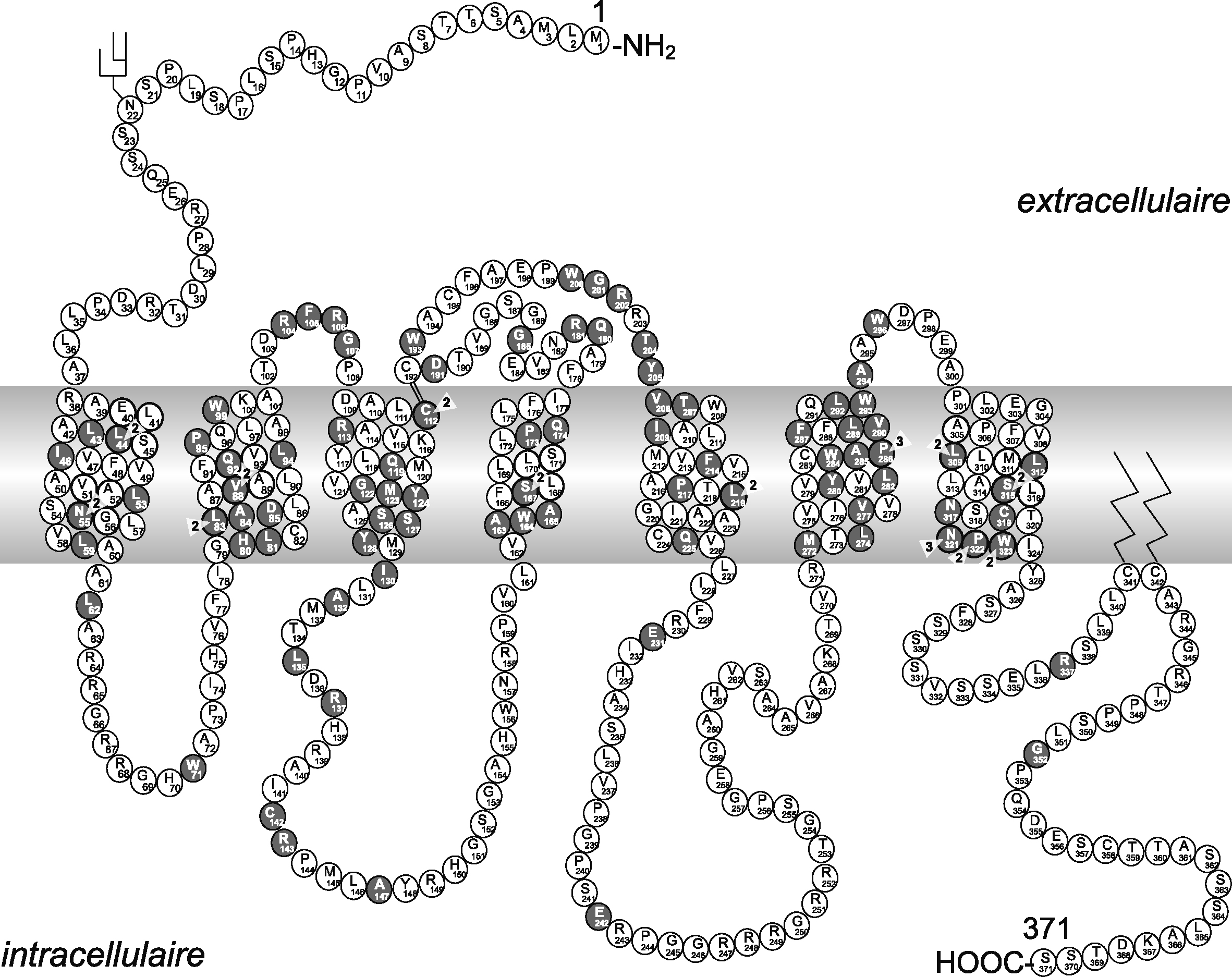
Représentation schématique de l'aquaporine 2 et de 35 mutations responsables de diabètes insipides néphrogéniques autosomiques dominants et récessifs.
Un monomère d'aquaporine est représenté avec six hélices transmembranaires. La localisation des boîtes NPA (asparagine, proline, alanine) est aussi représentée.
a Les chiffres entre parenthèses expriment les valeurs extrêmes observées. Ces données sont extraites de Documenta Geigy, Scientific Tables, 7th édition, 1970, p. 681.
Tableau 2
Débits urinaires (en ml/24 h) des nouveau-nés et des enfants
Les diabètes insipides néphrogéniques sont définis physiopathologiquement par l'impossibilité de concentrer maximalement les urines, et cliniquement par une polyurie. Ils peuvent être héréditaires ou secondaires à l'hypercalcémie, l'hypokaliémie ou l'administration de lithium. La résistance du tubule collecteur à la vasopressine (ou hormone antidiurétique) peut aussi être observée dans l'insuffisance rénale chronique et les néphropathies interstitielles. Les diabètes insipides néphrogéniques héréditaires peuvent être purs, c'est-à-dire exclusivement aquarétiques, ou complexes quand la diurèse pathologique observée s'accompagne d'une natriurèse et d'autres perturbations de l'excrétion électrolytique (potassium, calcium). Les diabètes insipides néphrogéniques héréditaires purs (aquarétiques) sont secondaires, soit à la perte de fonction du récepteur V2 antidiurétique de la vasopressine (AVPR2), soit à la perte de fonction du canal à l'eau dépendant de la vasopressine, l'aquaporine 2 (AQP2). Les mutations du gène AVPR2 (Xq28) sont responsables du diabète insipide néphrogénique lié à l'X. Les mutations du gène AQP2 (12p13) sont responsables du diabète insipide néphrogénique à transmission autosomique dominante ou récessive. Les diabètes insipides néphrogéniques héréditaires complexes (aquarétiques et natriurétiques) sont observés dans les syndromes de Bartter anténataux avec hyperprostaglandinurie et dans la cystinose. Le diagnostic génétique périnatal et le traitement précoce des enfants atteints de diabète insipide néphrogénique héréditaire permettent de prévenir et de traiter les épisodes sévères de déshydratation.
Nephrogenic diabetes insipidus which can be inherited or acquired, is characterized by an inability to concentrate urine despite normal or elevated plasma concentrations of the antidiuretic hormone, arginine–vasopressine (AVP). Polyuria, with hyposthenuria and polydipsia are the cardinal clinical manifestations of the disease. Hypercalcemia, hypokaliemia, lithium administration and chronic renal failure are the principal causes of acquired nephrogenic diabetes insipidus. About 90 percent of patients with congenital nephrogenic diabetes insipidus are males with X-linked recessive nephrogenic diabetes insipidus who have mutations in the arginine–vasopressin receptor 2 (AVPR2) gene that codes for the vasopressin V2 receptor. The gene is located in chromosome region Xq28. In about 10 percent of the families studied, congenital nephrogenic diabetes insipidus has an autosomal recessive or autosomal dominant mode of inheritance. In these cases, mutations have been identified in the aquaporin-2 gene (AQP2), which is located in chromosome region 12q13 and codes for the vasopressin-sensitive water channel. Other inherited disorders with mild, moderate or severe inability to concentrate urine include Bartter's syndrome and Cystinosis. Identification of the molecular defect underlying congenital nephrogenic diabetes insipidus is of immediate clinical significance because early diagnosis and treatment of affected infants can avert the physical and mental retardation associated with episodes of dehydration.