Médecine thérapeutique
MENUMaladie de Buerger : à propos d’un cas Volume 24, numéro 2, Mars-Avril 2018


La maladie de Buerger ou thromboangéite oblitérante (TAO) est une vasculopathie, non athéromateuse, occlusive et segmentaire touchant les artères et les veines de petits et moyens calibres, à l’origine de lésions ischémiques plus ou moins sévères, principalement de l’extrémité distale des membres supérieurs et inférieurs, et pouvant conduire dans certains cas à des amputations. L’étiologie précise de la maladie est inconnue, mais le tabac joue un rôle déterminant dans sa physiopathologie. Le diagnostic reste avant tout un diagnostic d’élimination. Il n’existe pas de traitement spécifique de la maladie, même si le sevrage complet et définitif du tabac en est la pierre angulaire. Nous rapportons ici un cas de nécrose bilatérale des orteils chez un patient tabagique, compatible avec le diagnostic de maladie de Buerger.
Le cas
Patient de 49 ans hospitalisé via les urgences pour un tableau de nécrose distale des deux pieds évoluant depuis quelques jours.
On note dans ses antécédents un tabagisme actif et une alcoolodépendance.Il est célibataire sans enfants, vit chez sa mère et exerce la profession de bûcheron.
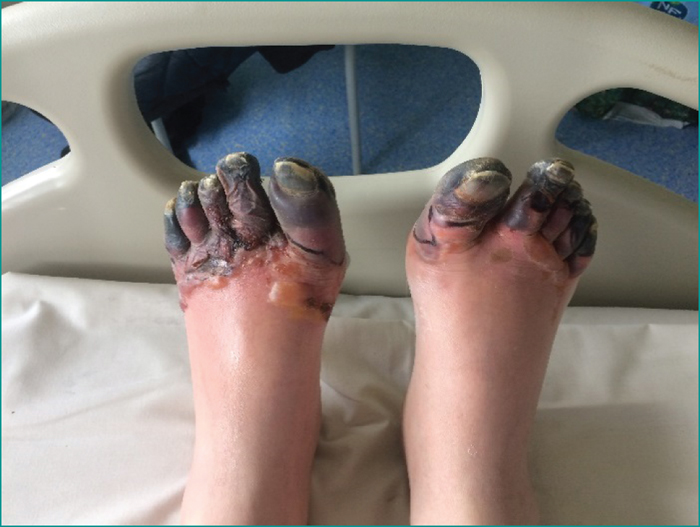
L’examen clinique initial met en évidence une nécrose complète de tous les orteils des deux pieds, avec quelques lésions phlycténulaires et un halo inflammatoire périphérique, ainsi que des lésions d’hyperkératoses plantaires (figure 1). Les pouls pédieux et tibiaux postérieurs sont bien perçus et il n’existe pas d’anomalie sensitivomotrice à l’examen neurologique. Il est subfébrile, à 38,2 ̊C, et l’hémodynamique est conservée.
Sur le plan biologique, il présente une hyperleucocytose à 16 000/mm3 (prédominance de polynucléaires neutrophiles), un syndrome inflammatoire biologique avec une protéine C réactive à 320 mg/L, une élévation des créatine phosphokinases (CPK) à 10 140 UI/L, sans insuffisance rénale associée.
Devant une forte suspicion d’infection des parties molles adjacentes, une antibiothérapie probabiliste par amoxicilline-acide clavulanique est instaurée, après la réalisation d’une série d’hémocultures qui resteront stériles.
Il bénéficie rapidement d’une échographie-Doppler artérielle des membres inférieurs qui montre :
- –une infiltration athéromateuse modérée, intéressant les artères fémorales communes sans sténose ni retentissement hémodynamique,
- –le flux triphasique est de bonne qualité au niveau des artères iliaques communes, iliaques externes, fémorales communes et fémorales superficielles, ainsi qu’au niveau des artères poplitées droites et gauches,
- –le flux artériel est satisfaisant au niveau des artères tibiales antérieures et postérieures, à droite comme à gauche,
- –l’aorte abdominale, terminale, présente un calibre normal et elle est parfaitement perméable.
L’échographie cardiaque transthoracique montre :
- –des cavités cardiaques de taille normale,
- –une bonne fonction du ventricule gauche, estimée à 71 %,
- –une insuffisance aortique minime sans autre valvulopathie,
- –pas de thrombus intracavitaire visible.
Un angioscanner des membres inférieurs est réalisé, qui ne met pas en évidence d’anomalie significative en dehors de calcifications athéromateuses discrètes de l’artère fémorale commune, un peu plus marquée à droite, mais sans sténose décelable. On note un aspect perméable des artères fémorales superficielles droite et gauche ainsi que du trépied jambier.
Un avis orthopédique est pris qui ne retient pas initialement l’indication d’une amputation.
Un avis en chirurgie vasculaire est également pris, évoquant en premier lieu une maladie de Buerger.
Il existe initialement une bonne évolution des signes inflammatoires locaux, avec stabilisation de la nécrose digitale, régression du syndrome inflammatoire biologique et des CPK.
Le bilan biologique est complété avec un bilan lipidique, qui revient normal, une hypoalbuminémie initiale à 23 g/L qui augmentera secondairement à 31 g/L après correction du syndrome inflammatoire et prise en charge nutritionnelle. Les facteurs antinucléaires (FAN) sont négatifs ainsi que les anticorps anti-antigènes nucléaires solubles. La recherche d’une cryoglobuline est négative, ainsi que les anticorps anti-cytoplasme des polynucléaires neutrophiles (ANCA). Le dosage du complément sérique retrouve un CH50 > 60 avec un C3 et un C4 normaux.
Après un mois de soins locaux, on note une momification de tous les orteils, avec une tendance à l’extension de la nécrose (figure 2) motivant finalement la décision d’amputation transmétatarsienne bilatérale.
L’anatomopathologie montre, à l’examen microscopique, des nécroses ischémiques massives de tous les plans tissulaires. Les structures artérielles encore visibles montrent un épaississement fibreux intimal non inflammatoire, des thrombi en cours d’organisation et, plus focalement, des aspects de surinfection. En conclusion, il s’agit de lésions d’ischémies massives et de surinfections, dans un contexte de vasculopathie peu spécifique mais compatible avec une maladie de Buerger.
Les suites opératoires seront simples avec une bonne cicatrisation.
L’arrêt du tabac est bien entendu fortement encouragé, avec consultation spécialisée.
Discussion
La maladie de Buerger appelé également thromboangéite oblitérante (TAO) consiste en une artériopathie non athéromateuse, segmentaire et inflammatoire, touchant de façon préférentielle le sujet jeune et atteignant les artères, mais également les veines de petit et de moyen calibre [1].
Elle n’est pas définie comme une vascularite proprement dite, du fait de sa forte association avec la consommation de tabac, mais également du respect relatif des parois vasculaires, de l’absence, habituellement notée, de signes généraux et de stigmates d’auto-immunité, ainsi que de la normalité des paramètres inflammatoires [2]. Dans notre observation, la présence d’un syndrome inflammatoire biologique à l’admission a été rattachée à une infection des parties molles, d’évolution favorable sous antibiothérapie.
Sur le plan épidémiologique, la prévalence de la maladie de Buerger est difficile à estimer, en raison d’une absence de critères spécifiques pour établir le diagnostic.
La répartition semble être ubiquitaire, même si l’on note une certaine prédominance au Moyen-Orient, en Asie et en Extrême Orient [3]. L’incidence a été estimée entre 8 et 11,6/100 000 personnes/an en Amérique du Nord [4], avec une baisse, rapportée par la Mayo Clinic, entre 1949 et 1986, avec 104 cas/100 000 admissions en 1949 versus 13/100 000 en 1986. Cette baisse peut être expliquée en partie par la diminution de la consommation de tabac, mais également par l’évolution des critères diagnostiques de la maladie. Cette tendance à la diminution de l’incidence de la TAO a été également rapportée par d’autres auteurs en Pologne, Europe de l’Ouest, Asie [5], mais pas dans d’autres régions, et notamment au Moyen-Orient [1].
La maladie prédomine chez l’homme, même si le rapport homme/femme tend à se réduire, ce qui peut s’expliquer en partie par une augmentation importante de la consommation de tabac chez les femmes [3, 5, 6]. C’est une maladie de l’adulte jeune : dans une série américaine portant sur 112 patients entre 1970 et 1987, la moyenne d’âge était de 42 ans avec des extrêmes de 20 et de 75 ans, et 7 % des patients de plus de 60 ans [3].
Parmi les hypothèses physiopathologiques, l’intoxication tabagique joue un rôle central dans l’initiation et l’évolution de la maladie [1]. Bien qu’un article de la littérature ait rapporté un cas de maladie de Buerger chez un patient non tabagique, il faut considérer qu’il n’y a pas de TAO sans tabagisme actif [7]. Une étude indienne suggère une relation dose-dépendante avec un odds-ratio de 58,8 chez les sujets fumant de plus de vingt cigarettes par jour, par rapport à ceux en consommant moins de dix par jour [5]. Des atteintes similaires sont notées chez des utilisateurs de cannabis – sachant que cette consommation est fréquemment associée à celle du tabac [3].
Sur le plan génétique, l’hypothèse d’une prédisposition à développer ou non la maladie a été évoquée, devant l’hétérogénéité de sa répartition géographique. Des études ont noté un haplotype HLA qui semble favorisant, mais de manière peu significative et surtout différemment selon les pays étudiés [2, 3, 5].
L’auto-immunité a été également explorée. Ainsi, une réactivité plus importante des lymphocytes vis-à-vis des collagènes de type I et III, constituants des parois vasculaires, a été rapportée chez les patients porteurs de la maladie de Buerger, ainsi que la présence d’anticorps anticollagène. Cependant, ces résultats sont considérés comme non spécifiques et n’ont pas été confirmés [2]. Des résultats contradictoires existent également sur la présence d’ANCA [5].
La présence sur les pièces anatomopathologiques de thrombi au sein des vaisseaux atteints (constatée chez notre patient) a conduit à rechercher l’existence d’une thrombophilie sous-jacente. Une étude, portant sur un effectif faible de patients (n = 36), a rapporté, chez trente-cinq d’entre eux, une mutation G20210A du gène de la prothrombine, dont vingt-neuf à l’état d’homozygotie, sans anomalie notée dans le groupe contrôle [8]. Cependant, ces données n’ont pu être confirmées par une autre étude dans laquelle le bilan de thrombophilie était plus exhaustif [9].
Enfin, l’endothélium vasculaire semble également jouer un rôle important dans la physiopathologie de la maladie, par le biais d’un trouble de la relaxation et par l’augmentation de l’expression des molécules d’adhésion [1, 3, 5].
En définitive, l’étiologie précise de la maladie de Buerger reste à ce jour inconnue. Elle est vraisemblablement multifactorielle, associant prédisposition génétique, rôle du tabac, réaction auto-immune, trouble de l’hémostase et réactivité des cellules endothéliales vasculaires.
La présentation clinique de la maladie de Buerger se caractérise principalement par des atteintes artérielles, mais aussi veineuses, des membres. Ainsi, la claudication de la plante du pied est un signe fréquent de la maladie et relativement précoce, allant de 44 % [7] à 63 % [5] selon les séries. Cette claudication peut s’étendre au mollet au cours de l’évolution de la maladie. Un phénomène de Raynaud peut être présent, variant de 2 à 44 % selon les études [1, 2, 5]. Les membres inférieurs sont atteints dans 100 % des cas, contre 44 % pour les membres supérieurs [5]. Dans la série d’Olin [1] les troubles trophiques étaient présents dans 76 % des cas au moment du diagnostic, contre 7 % dans la série de Malecki [5]. Les atteintes veineuses consistent principalement en des thrombophlébites superficielles et migratrices (40 à 60 % des cas), touchant les membres supérieurs et inférieurs, et dont l’évolution semble parallèle à celle de l’activité de la maladie [2]. De rares atteintes artérielles atypiques de siège aorto-iliaques [5], mais touchant aussi les artères digestives, cérébrales et coronaires [2], ont été rapportées. Des atteintes rénales sont également notées [15].
Parmi les atteintes extravasculaires, on peut observer des atteintes articulaires faites de monoarthrites aiguës migratrices et non érosives, touchant de façon préférentielle les genoux et les poignets et pouvant précéder les signes ischémiques, de dix ans parfois, dans environ 12,5 % des cas [2]. De façon plus anecdotique, ont été publiés dans la littérature des cas de spondylarthropathies HLA B27 positives [2], de polyarthrites et de syndrome du canal carpien [3].
Une atteinte nerveuse est également suspectée, devant des manifestations de type paresthésies, décrites dans 15 à 69 % des cas selon les séries [1, 5]. Ces atteintes pourraient être liées à une neuropathie ischémique ou à une atteinte inflammatoire du paquet nerveux.
Sur le plan biologique, la normalité des paramètres est habituellement la règle et constitue un des éléments du diagnostic positif de la maladie de Buerger. Les paramètres inflammatoires sont normaux, sauf en cas d’infection des parties molles associée, comme c’est le cas dans notre observation. Sur le plan immunologique, les ANCA sont négatifs, ainsi que les FAN. Le bilan doit comprendre les tests d’hémostase, dont la recherche d’anticorps antiphospholipides [7], qui n’ont malheureusement pas été recherchés chez notre patient.
L’échographie cardiaque (par voie transthoracique et/ou transœsophagienne) permet d’éliminer une cause embolique d’origine cardiaque, ce qui est le cas dans notre observation.
L’artériographie, quand elle est réalisée, ne montre pas de signe pathognomonique de la maladie [3]. On note une atteinte distale des artères de moyen et de petit calibre, volontiers segmentaire, et sans élément orientant vers une athérosclérose, ni pour une origine embolique. L’aspect classique en « tire-bouchon » (cork-screw des Anglo-Saxons, ou signe de Martonel) correspond à une dilatation des artères collatérales et peut se rencontrer dans toutes les atteintes artérielles distales (lupus systémique, CRESTi-syndrome, vascularite rhumatoïde, syndrome des antiphospholipides, syndrome de Sharp) et n’a donc rien de spécifique [1].
Les lésions anatomopathologiques, rarement réalisées, montrent une atteinte segmentaire et plurifocale, touchant les artères et les veines des extrémités distales, de petit et moyen calibre (1-5 mm de diamètre). Il n’est pas retrouvé de nécrose fibrinoïde et pas d’atteinte de la limitante élastique interne, contrairement à la maladie athéroscléreuse ou aux vascularites systémiques. À la phase aiguë on retrouve des thromboses souvent occlusives.
En l’absence de marqueur spécifique de la maladie de Buerger, de nombreux critères diagnostiques ont été élaborés, basés à la fois sur des critères d’exclusion et des éléments cliniques et artériographiques compatibles avec le diagnostic [2]. Ce sont notamment le score d’Adar [16], les critères de Mills et Porter [17], de Papa [18], de Shionaya [19] et d’Olin [1], qui sont les plus récents et dont l’utilisation en pratique est recommandée [5, 10]. Ces critères comprennent un âge de survenue avant 45 ans (49 ans pour notre patient), un tabagisme actif (présent chez notre patient), des symptômes d’ischémie distale des membres (présents chez notre patient), des paramètres biologiques éliminant d’autres étiologies comme une pathologie auto-immune type vascularite, un diabète (absent chez notre patient), l’exclusion d’une source embolique (échographie cardiaque transthoracique normale chez notre patient) et des données artériographiques compatibles [1]. L’artériographie n’a pas été réalisée chez notre patient, car l’examen n’est pas disponible dans notre établissement ; elle a été remplacée par un angioscanner artériel des membres inférieurs, qui a permis d’éliminer une cause athéromateuse.
Le pronostic de la maladie de Buerger est avant tout local et fonctionnel, lié au risque d’amputation. Dans une série de 111 patients suivis pendant 15,6 ans, le risque d’amputation était de 25 % à cinq ans, de 38 % à dix et de 46 % à vingt. Un lien très fort est rapporté avec la poursuite ou non de l’intoxication tabagique. Ainsi, dans cette étude, le risque d’amputation disparaissait huit ans après le sevrage définitif du tabac, avec un taux d’amputation de 84,3 % en cas de poursuite de l’intoxication contre 30,6 % en cas d’arrêt du tabac [20]. Des résultats similaires ont été obtenus dans une étude japonaise portant sur près de 850 patients, avec un risque d’amputation 2,73 fois plus élevé chez les patients qui poursuivaient leur intoxication tabagique [21]. Classiquement on ne décrit pas d’augmentation de la morbimortalité dans la, maladie de Buerger, avec une survie de 90 à 95 % à dix ans et de 85 % à vingt-cinq [2]. L’étude de Cooper et al.[4] rapporte néanmoins une augmentation de la mortalité, laquelle reste élevée même après l’arrêt du tabac et lors du suivi à long terme, avec notamment 75 % de troubles métaboliques à vingt ans (intolérance au glucose et diabète, dyslipidémie, hyperuricémie), et 13,6 % d’atteinte artérielle proximale typique d’athérosclérose en cas de suivi supérieur à huit ans.
Le seul traitement ayant prouvé son efficacité dans le traitement de la maladie de Buerger, est l’arrêt complet et définitif du tabac [1].
La prise en charge locale des plaies, afin de prévenir les surinfections et permettre la cicatrisation, est un élément majeur de la prise en charge, et ce d’autant plus s’il existe des troubles trophiques [2, 3].
L’ilomédine (Iloprost®), analogue de la prostacycline par voie intraveineuse, a été comparé à l’aspirine dans une étude multicentrique européenne, chez 152 patients atteints de maladie de Buerger [11]. Au terme des vingt-huit jours de traitement, la cicatrisation des lésions chez les sujets présentant des troubles trophiques était obtenue dans 35 % des cas sous ilomédine versus 13 % sous aspirine (p = 0,05). Ces résultats prometteurs n’ont pas été confirmés par d’autres études, notamment contre placebo. Plus récemment, une étude turque, ouverte, prospective et portant sur 150 patients a montré une efficacité de 60,2 % sur la guérison complète des ulcères initiaux, en comparaison aux troubles trophiques à l’inclusion des patients (p < 0,001) [12]. Il paraît actuellement peu probable que des études plus significatives, avec un design différent (études de supériorité et a fortiori contre placebo), soient un jour réalisées, compte tenu du peu de traitements disponibles dans cette pathologie.
Le bien-fondé de la sympathectomie dorsale ou lombaire est controversé [3]. Une étude a même montré son infériorité par rapport à l’ilomédine [14].
Une étude non randomisée et non contrôlée, portant sur un effectif de douze patients seulement, a utilisé le bosentan, antagoniste des récepteurs de l’endothéline-1 et puissant vasoconstricteur (ayant prouvé son efficacité dans l’hypertension artérielle pulmonaire et dans la prévention des récidives d’ulcères dans la sclérodermie systémique), à la posologie initiale de 62,5 mg deux fois par jour pendant un mois, puis 125 mg deux fois par jour, avec un suivi médian de vingt mois [13]. Sur le plan clinique, sur treize extrémités, douze ulcères étaient améliorés, deux amputations étaient nécessaires, et une amélioration des flux artériels était observée chez dix des douze patients, aussi bien en artériographie qu’en IRM. Cependant, ces résultats, peu significatifs, demandent à être vérifiés par d’autres études.
Le traitement chirurgical au cours de la maladie de Buerger consiste principalement en des amputations, comme c’est malheureusement le cas dans notre observation. Les revascularisations ne sont que rarement réalisées en raison du caractère très distal et diffus des lésions vasculaires, associé à un fort taux de ré-occlusions des pontages à ce niveau et de la courte vie du greffon, même si des études ont montré une guérison des ulcères cutanés dans 91 % des cas en cas de greffe [5]. D’autres techniques chirurgicales doivent encore être évaluées comme la greffe omentale [22].
Conclusion
La maladie de Buerger, ou thromboangéite oblitérante, est une vasculopathie artérioveineuse non athéromateuse du sujet jeune et fumeur, touchant les vaisseaux de petit et moyen calibre, à l’origine de lésions ischémiques plus ou moins graves, localisées essentiellement à l’extrémité distale des membres supérieurs et inférieurs, et pouvant nécessiter, dans un certain nombre de cas, des gestes d’amputations. L’étiologie précise est inconnue et la physiopathologie est multifactorielle avec un rôle prépondérant du tabac. Il n’existe pas de traitement spécifique de la maladie, en dehors de l’arrêt complet et définitif de l’intoxication tabagique. Les analogues de la prostacycline intraveineuse (ilomédine) se sont révélés prometteurs dans certaines études, lesquelles n’ont toutefois pas été confirmées notamment par des observations contre placebo. La chirurgie d’amputation est parfois nécessaire, comme nous le rapportons dans notre observation, avec des conséquences fonctionnelles parfois difficiles.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt en rapport avec cet article.
i Calcifications sous-cutanées (C), syndrome de Raynaud (R), anomalies œosophagiennes (E), sclérodactylie (S), télangiectasies (T).
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International