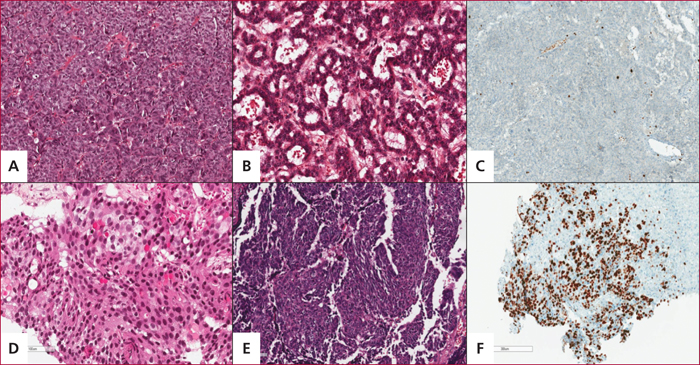
Aspects histologiques des tumeurs neuroendocrines pancréatiques bien différenciées d’architecture compacte (A) ou microkystique (B) avec un index de prolifération bas (Ki67<1%, soit grade 1) (C), et des carcinomes neuroendocrines peu différenciés à grandes (D) ou à petites cellules (E) avec un index de prolifération élevé (60 %, soit grade 3) (F) (Dr J. Cros, Hôpital Beaujon).
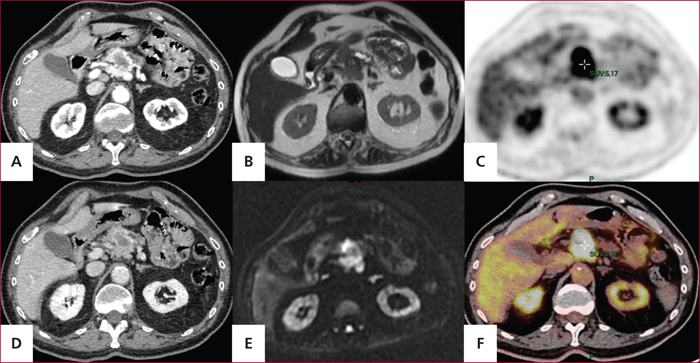
Tumeur neuroendocrine pancréatique bien différenciée de grade 2 (Ki67 18 %) associé à des métastases ganglionnaires, chez un patient de 68 ans. Aspect en tomodensitométrie avec injection de contraste aux temps artériel précoce (A) et portal (D), en IRM pondérée en T2 (B) en IRM de diffusion (D), et en tomographie par émission de positons au 18FDG (C et F).
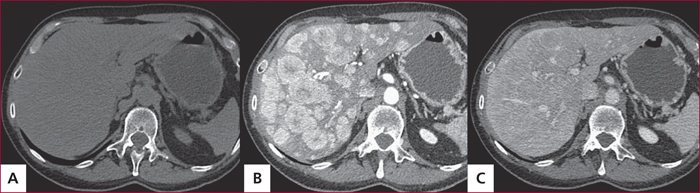
Tomodensitométrie hépatique sans injection de produit de contraste (A) et aux temps artériel (B) et portal (C), permettant le bilan des MH. Celles-ci peuvent ne pas être visualisées en l’absence d’acquisition temps artériel précoce (Dr M. Zappa, Hôpital Beaujon).
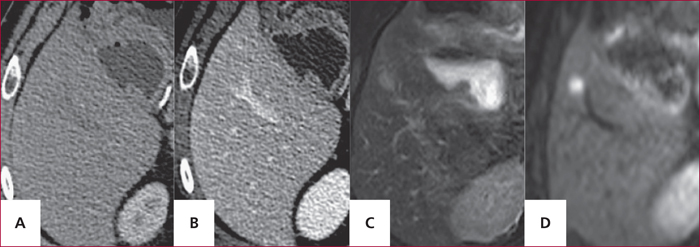
Tomodensitométrie hépatique aux temps artériel (A) et portal (B) et IRM hépatique pondérée en T2 (C) et en diffusion (D), réalisés dans le cadre du bilan d’extension d’une TNEP. Mise en évidence sur l’IRM de diffusion d’une métastase non visible en tomodensitométrie (Dr M. Zappa, Hôpital Beaujon).
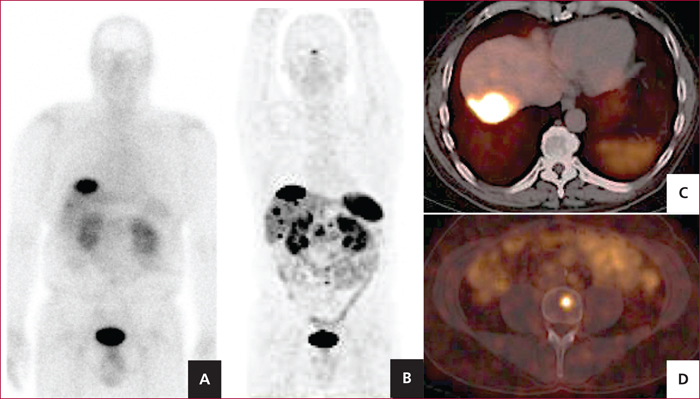
Scintigraphie des récepteurs de la somatostatine au 111In-pentetreotide (A) réalisée dans le cadre du bilan d’extension d’une TNEP, montrant une métastase du dôme hépatique. En comparaison, la TEP au 68Ga-DOTATOC montre l’existence de multiples MH (B) en plus de celle du dôme (C), ainsi qu’une localisation rachidienne L4 (D).
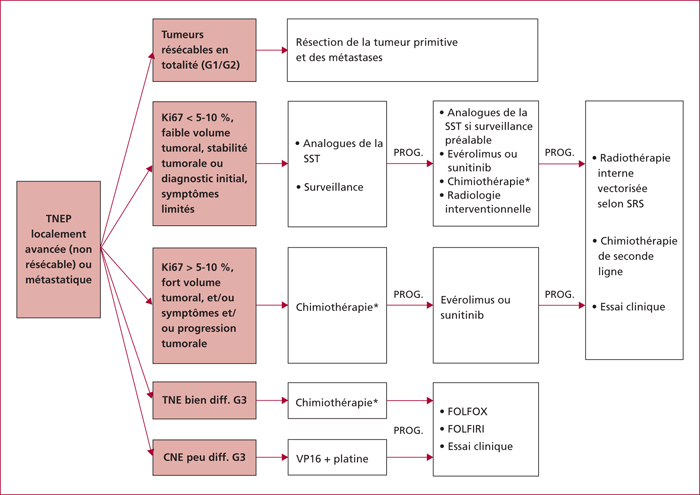
Algorithme de prise en charge thérapeutique des TNEP localement avancées ou métastatiques selon l’ENETS (adapté d’après [35]).
*Les chimiothérapies recommandées en première ligne sont les associations streptozotocine-doxorubicine, streptozotocine-5-fluorouracile et témozolomide-capécitabine. CNE, carcinome neuroendocrine ; SRS, scintigraphie des récepteurs de la somatostatine ; SST, somatostatine ; TNEP, tumeur neuroendocrine pancréatique.
Les tumeurs neuroendocrines pancréatiques (TNEP) sont des tumeurs rares, mais dont l’incidence augmente. Elles sont caractérisées par un fort taux de métastases (50 %), principalement hépatiques. Du fait de leur croissance tumorale relativement lente, elles sont néanmoins associées à des taux de survie prolongée (environ 50 % à 5 ans).
Les TNEP sont le plus souvent sporadiques et diagnostiquées fortuitement ou devant des signes cliniques aspécifiques en lien avec les localisations tumorales. Moins de 20 % des TNEP sont fonctionnelles et sont responsables de symptômes liés à l’hypersecrétion d’une hormone qui doivent être traités en priorité. Le bilan d’extension comporte le dosage plasmatique de la chromogranine A, un scanner et/ou une IRM abdominale et une imagerie nucléaire des récepteurs de la somatostatine voire une TEP au FDG. Les principaux facteurs pronostiques sont la différenciation et le grade histologiques, le stade tumoral, la pente évolutive et le volume tumoral, notamment hépatique.
Le traitement des TNEP localisées repose sur la résection chirurgicale, à l’exception de certains incidentalomes sporadiques < 2 cm. Les options thérapeutiques des TNEP bien différenciées associées à des métastases non résécables incluent la chirurgie, la chimiothérapie, les thérapies ciblées (sunitinib, everolimus), les analogues de la somatostatine, la (chimio)-embolisation hépatique et la radiothérapie interne vectorisée.