Hépato-Gastro & Oncologie Digestive
MENUPrise en charge thérapeutique des cancers colorectaux métastatiques au-delà de la deuxième ligne Volume 26, numéro 3, Mars 2019

Illustrations
Tableaux
La survie globale des patients atteints d’un cancer colorectal métastatique atteint ou dépasse actuellement 30 mois en médiane [1-3]. L’amélioration de cette survie globale au cours des trois dernières décennies est corrélée au nombre croissant de molécules disponibles, à la diffusion et aux progrès du traitement chirurgical des métastases, auquel il faut ajouter les avancées des traitements de thermo-ablation (par voie chirurgicale ou non) [4]. Dans ce contexte, jusqu’à 40 % des patients reçoivent actuellement une troisième ligne de traitement [2, 3, 5].
Les études de phase III internationales CORRECT, RECOURSE, mais aussi CONCUR et TERRA (études restreintes au continent asiatique), ont validé, chez 2 170 patients au total, l’intérêt du régorafénib et du trifluridine/tipiracil chez les patients avec cancer colorectal métastatique en échec des traitements standards (fluoropyrimidines, oxaliplatine, irinotécan, bévacizumab et, en cas de tumeur RAS sauvage, anti-EGFR) [6-9]. Ces deux chimiothérapies sont devenues, à la suite des résultats de ces études, deux traitements standards validés au-delà de la deuxième ligne thérapeutique.
Dans cet article, nous résumerons les données pharmacocinétiques importantes, les données d’efficacité et de tolérance ayant conduit à l’enregistrement de ces deux médicaments, les données actuelles concernant la recherche de biomarqueurs prédictifs de leur efficacité, ainsi que les autres options thérapeutiques envisageables au-delà de la deuxième ligne. Le régorafénib et le triflirudil/tipiracil sont deux standards validés au-delà de la deuxième ligne dans la prise en charge du cancer colorectal métastatique
Pharmacocinétique
Le trifluridine/tipiracil et le régorafénib ont une administration orale. Les questions relatives aux troubles potentiels de leur absorption ou à d’éventuelles interactions médicamenteuses reviennent de ce fait à l’ordre du jour. Ces deux molécules présentent des particularités pharmacocinétiques et pharmacodynamiques distinctes.
Trifluridine/tipiracil
Le trifluridine/tipiracil se compose de trifluridine et de chlorhydrate de tipiracil (rapport molaire de 1:0,5). La concentration plasmatique de la trifluridine diminue rapidement avec une demi-vie extrêmement courte, d’environ 12 minutes. Chez l’homme, la trifluridine est dégradée par la thymidine phosphorylase en produits inactifs. Le chlorhydrate de tipiracil, inhibiteur compétitif de la thymidine phosphorylase, inhibe la dégradation de la trifluridine [10]. Il augmente ainsi la concentration de la trifluridine, qui va agir au sein du noyau en inhibant la réplication de l’ADN, d’où découle son activité anti-tumorale. Aucune dégradation n’est effectuée par la dihydropyrimidine déshydrogénase, enzyme de dégradation du 5-fluoro-uracile, ce qui permet d’envisager l’utilisation du trifluridine/tipiracil chez les patients présentant un déficit, notamment complet, en cette enzyme. Le trifluridine/tipiracil peut être utilisé en cas de déficit, y compris complet, en dihydropyrimidine déshydrogénase, enzyme de dégradation du 5-FU, qui n’intervient pas dans son catabolisme
Dans une étude de phase I, chez des patients japonais atteints de tumeurs solides et recevant le trifluridine/tipiracil, la concentration plasmatique maximale (Cmax) de la trifluridine a augmenté de 2,6 fois au douzième jour (J) par rapport au J1, alors que les dosages pharmacocinétiques du chlorhydrate de tipiracil à J12 étaient identiques à ceux à J1 [11]. Sur la base des résultats pharmacologiques, la posologie retenue de trifluridine/tipiracil est de 70 mg/m2/jour (35 mg/m2/prise 2 fois/j). La prise du trifluridine/tipiracil pendant un repas riche en graisses et en calories entraîne une baisse des Cmax de la trifluridine et du chlorhydrate de tipiracil d’environ 40 % par rapport à une prise à jeun (à 1 heure du dernier repas) [10]. Ainsi, il convient de privilégier une administration à au moins 1 heure du dernier repas.
L’étude de phase III RECOURSE a mis en évidence une toxicité principalement hématologique du trifluridine/tipiracil, liée au pic de concentration et à la demi-vie courte du produit [7]. Comme indiqué précédemment, le pic plasmatique à J12 de la trifluridine est 2,6 fois supérieur au pic constaté au J1 du traitement. Ainsi, les toxicités hématologiques sont principalement observées pendant la deuxième séquence du cycle. La réduction du nombre de neutrophiles a été montrée comme significativement corrélée à la Cmax et à l’aire sous la courbe de concentration plasmatique de la trifluridine [11]. Pour autant, il n’est pas encore possible à ce jour, en routine, de prévenir ou diminuer les toxicités, notamment la toxicité hématologique du trifluridine/tipiracil, grâce à une adaptation pharmacocinétique individuelle. L’utilisation et les modalités optimales d’administration des G-CSF dans cette situation restent débattues. Le trifluridine/tipiracil s’administre per os au moins une heure après la fin du repas à la dose de 35 mg/m2 deux fois par jour de J1 à J5 puis de J8 à J12 tous les 28 jours. Une adaptation pharmacocinétique individuelle de sa posologie n’est pas possible en routine
Régorafénib
Le régorafénib est un inhibiteur oral de plusieurs protéines kinases, impliquées notamment dans la régulation de l’angiogenèse tumorale (VEGFR-1, -2 et -3 et Tie-2), l’oncogenèse (KIT, RET, RAF-1, BRAF [y compris dans sa forme mutée V600E]) et le microenvironnement tumoral (PDGFR et FGFR) [12]. Des essais de phase III ont démontré une activité anti-tumorale du régorafénib dans plusieurs tumeurs solides : le carcinome hépatocellulaire avancé, les tumeurs stromales gastro-intestinales métastatiques et le cancer colorectal métastatique [6, 13, 14].
Le régorafénib étant administré par voie orale, la caractérisation de ses voies de biotransformation et de son activité métabolique revêt une importance particulière. Chez l’homme, le régorafénib est principalement métabolisé dans le foie. Il y subit une biotransformation oxydative et conjuguée, conduisant à la formation du principal métabolite N-oxyde M-2. Le métabolite M-2 passe dans la circulation systémique. Une biotransformation oxydative supplémentaire conduit à un métabolite secondaire, le dérivé N-oxyde déméthylé M-5 [12]. Des essais cliniques chez des patients atteints de cancer avancé ont démontré que les métabolites plasmatiques M-2 et M-5 ont une activité inhibitrice de tyrosines kinases [15-17]. Les métabolites M-2 et M-5 contribuent ainsi à l’activité clinique globale du régorafénib. Le régorafénib et ses deux métabolites M-2 et M-5 présentent des concentrations inhibitrices (IC50) différentes. Par exemple, l’IC50 du VEGFR-2 est de 40, 30 et 20 nM respectivement avec le régorafénib, le métabolite M-2 et le métabolite M-5 [12]. De plus, la demi-vie du régorafénib et du métabolite M-2 est de 24 heures, contre une semaine pour le métabolite M-5. Enfin, des études in vitro ont montré une forte liaison aux protéines plasmatiques du régorafénib et de ses métabolites M-2 et M-5, avec des fractions plasmatiques libres chez l’homme d’environ 0,5 %, 0,2 %, et 0,05 %, respectivement [12]. En conséquence des différences de fixation aux protéines, de capacité d’inhibition des protéines kinases et de demi-vies entre le régorafénib et chacun de ses deux métabolites, aucune relation entre le dosage plasmatique du régorafénib et son efficacité n’a été mise en évidence à ce jour.
L’étude ReDOS est une étude de phase II randomisée comparant chez 123 patients un schéma d’escalade de dose individuelle du régorafénib (80 mg/jour en semaine 1, puis 120 mg/jour en semaine 2 en l’absence de toxicité significative, puis 160 mg/jour, dose cible, en semaine 3 si la tolérance continue à rester satisfaisante) au schéma d’administration standard (160 mg/jour d’emblée, trois semaines sur quatre) dans le cancer colorectal métastatique [18]. Les résultats de cette étude montrent qu’une meilleure gestion de la toxicité durant les deux premiers cycles de régorafénib avec une stratégie d’augmentation progressive de la dose a conduit à une toxicité moindre, à une proportion significativement plus grande de patients débutant un troisième cycle de régorafénib (objectif principal de l’étude) (43 % vs. 24 %, p = 0,028) et à une survie globale numériquement supérieure, n’atteignant toutefois pas la significativité statistique dans cette étude de faible effectif (9,0 mois vs. 5,9 mois ; p = 0,09). Ces résultats plaident pour la diffusion de cette stratégie d’augmentation de dose mais n’affranchissent pas le clinicien d’une surveillance rapprochée pendant les deux premiers mois de traitement. Les résultats de l’essai de phase II randomisé RE-ARRANGE, évaluant deux schémas d’administration du régorafénib (120 mg/jour trois semaines sur quatre ou 160 mg/jour une semaine sur deux au cycle 1, puis 160 mg/jour 3 semaines sur 4 à partir du cycle 2) en sus du schéma standard ne sont pas encore connus. Le régorafénib s’administre per os d’emblée à la dose de 160 mg/jour, 3 semaines sur 4 ou mieux, en augmentant progressivement sa posologie en fonction de la tolérance à respectivement 80, 120, puis 160 mg/jour aux semaines 1, 2 et 3
Efficacité
L’étude de phase III RECOURSE a randomisé 800 patients avec cancer colorectal métastatique en échec des fluoropyrimidines, de l’oxaliplatine, de l’irinotécan, du bévacizumab, des anti-EGFR (en cas de tumeur RAS sauvage) et éventuellement, du régorafénib, au cours d’au moins deux lignes de traitement [7](tableau 1). Ils étaient randomisés selon un ratio 2:1 entre trifluridine/tipiracil (35 mg/m2 matin et soir 5 jours sur 7, 2 semaines sur 4, J1 = J28 ; n = 534) et placebo (n = 266) jusqu’à progression de la maladie ou apparition d’une toxicité inacceptable, associés dans les deux bras aux meilleurs soins de support. Les évaluations tumorales étaient réalisées toutes les 8 semaines selon les critères RECIST 1.1. Les critères secondaires incluaient l’impact du statut tumoral KRAS sur le traitement et le temps jusqu’à détérioration définitive du statut de performance (PS) (≥ 2).
La survie globale (critère de jugement principal) a été significativement améliorée dans le bras trifluridine/tipiracil (7,1 vs. 5,3 mois ; ratio de risque [HR] : 0,68 ; intervalle de confiance à 95 % [IC] : 0,58-0,81 ; p < 0,001) (tableau 1), de même que la survie sans progression et le taux de contrôle tumoral, mais non le taux de réponse objective tumorale, critères de jugement secondaires.
L’étude de phase III CORRECT a randomisé 760 patients avec cancer colorectal métastatique là aussi en échec des fluoropyrimidines, de l’oxaliplatine, de l’irinotécan, du bévacizumab et des anti-EGFR (en cas de tumeur RAS sauvage) – ici en revanche, les patients n’avaient pas reçu antérieurement de trifluridine/tipiracil [6](tableau 1). Ils étaient randomisés selon un ratio 2:1 entre régorafénib (160 mg/jour en une prise 3 semaines sur 4, J1 = J28 ; n = 505) et placebo (n = 255) jusqu’à progression de la maladie ou apparition d’une toxicité inacceptable, associés dans les deux bras aux meilleurs soins de support. Les évaluations tumorales étaient réalisées toutes les 8 semaines selon les critères RECIST 1.1.
La survie globale (critère de jugement principal) a été significativement améliorée dans le bras régorafénib (6,4 vs. 5,0 mois ; HR : 0,77 ; IC : 0,64-0,94 ; p = 0,0052) (tableau 1), de même que la survie sans progression et le taux de contrôle tumoral, mais ici encore pas le taux de réponse objective tumorale, critères de jugement secondaires.
Globalement on peut retenir de la comparaison indirecte du trifluridine/tipiracil et du régorafénib, d’après les résultats des deux essais de phase III d’enregistrement RECOURSE et CORRECT, les points-clés suivants :
- –populations de patients globalement comparables (plus de patients asiatiques [34 % vs. 15 %], de patients en quatrième ligne ou au-delà [60 % vs. 49 %] et de patients résistant aux fluoropyrimidines [98 % vs. 83 %] dans l’essai évaluant le trifluridine/tipiracil) ;
- –bénéfice de survie sans progression (HR : 0,48 vs. 0,49) et de survie globale (HR : 0,68 vs. 0,77) similaire ;
- –durée médiane de traitement identique (1,7 mois) ;
- –pourcentages de dose reçue/planifiée proches (89 % vs. 79 %) ;
- –absence de biomarqueurs cliniques identifiés, avec un bénéfice dans tous les sous-groupes analysés ;
- –profil de toxicité différent (voir chapitre suivant) principalement hématologique et digestive pour le trifluridine/tipiracil ; principalement toxicité cutanéo-muqueuse, hypertension artérielle et asthénie pour le régorafénib.
La comparaison indirecte du trifluridine/tipiracil et du régorafénib, à partir des études d’enregistrement de phase III, montre un bénéfice similaire de survie sans progression et de survie globale
Toxicité
Un élément-clé du choix thérapeutique au-delà de la deuxième ligne du cancer colorectal métastatique est le profil de toxicité des différents traitements à disposition. Ce profil sera à prendre en compte au regard des toxicités des traitements antérieurs (cytopénies, etc.), de la préférence du patient qui en aura été informé et des éventuelles contre-indications à chacun des traitements.
Régorafénib
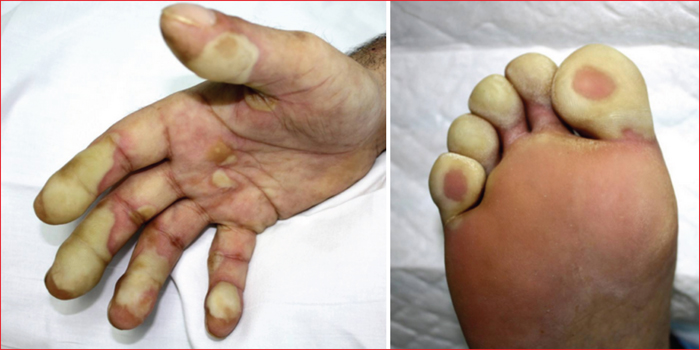
Dans les deux études de phase III CORRECT et CONCUR comparant le régorafénib au placebo, le régorafénib était associé à un taux de toxicité de grade 3-4 supérieur à 50 % [6, 8]. Les deux principaux effets indésirables limitants sont le syndrome main-pied et la diarrhée, le plus souvent de grade 1-2, mais pouvant être plus sévères dans 8-17 % des cas (tableau 2). Le syndrome main-pied se caractérise par l’apparition, 8 à 10 jours après la mise en route du traitement, d’une hyperkératose circonscrite, digitale et plantaire, sur les zones de pression et d’appui (figure 1). La fatigue, la perte d’appétit, la mucite et le rash cutané sont également fréquents. Sur le plan biologique, les anomalies les plus fréquentes sont la cytolyse hépatique, l’hyperbilirubinémie et l’hyperglycémie. Les autres effets indésirables rapportés sont peu fréquents (troubles du transit, douleurs abdominales, dysgueusie, alopécie, myalgies, sécheresse cutanée, hypophosphorémie…). En particulier, la toxicité hématologique sévère est rare. Enfin, le principal effet indésirable anti-angiogénique du régorafénib est l’hypertension artérielle, de grade 3 ou 4 dans 7-11 % des cas. Elle ne constitue pas en soi une contre-indication au traitement. Elle nécessite une surveillance régulière par auto-mesure à domicile, permettant de s’affranchir de l’hypertension artérielle surajoutée (effet « blouse blanche »). Les autres effets indésirables anti-angiogéniques rapportés sont peu fréquents (épistaxis, protéinurie, dysphonie).
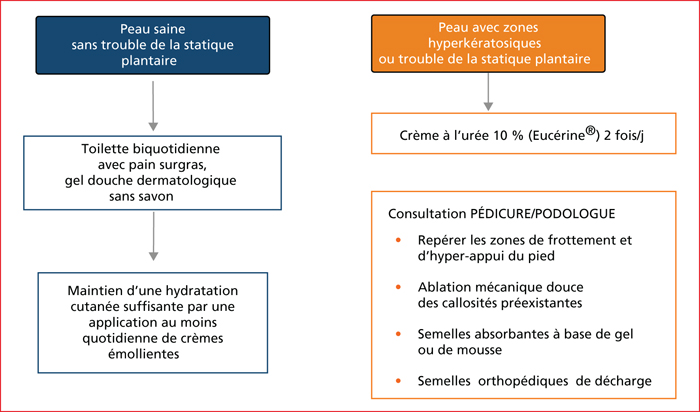
Le syndrome main-pied, fréquent et parfois invalidant lorsque sévère, doit faire l’objet d’une surveillance clinique rapprochée et d’une prise en charge spécifique intégrant des mesures préventives. Celles-ci visent à éviter la sécheresse cutanée, maintenir une hydratation cutanée suffisante, traiter les zones hyperkératosiques déjà présentes et améliorer d’éventuels troubles de la statique plantaire (figure 2)[19, 20]. Le traitement repose sur l’application alternée de topiques comportant des crèmes émollientes et cicatrisantes et de dermocorticoïdes qui pourront être associés à des bains tièdes. Y seront ajoutés des kératolytiques sur les zones d’hyperkératose (tableau 3).
La diarrhée ne relève pas d’une prise en charge préventive spécifique (hormis l’information et l’éducation du patient et le maintien d’une bonne hydratation). Elle doit faire simplement l’objet d’une ordonnance anticipatoire de lopéramide.
Concernant l’hypertension artérielle, une surveillance régulière et rapprochée est préconisée durant les six premières semaines de traitement, période durant laquelle survient cet effet secondaire. Un traitement anti-hypertenseur est à instaurer en monothérapie puis (si insuffisant) en bithérapie, avec une surveillance tensionnelle régulière. En cas d’échec du traitement anti-hypertenseur, la dose de régorafénib doit être diminuée de 40 mg/jour.
Vis-à-vis de la cytolyse hépatique, une interruption du régorafénib doit être réalisée en cas d’ASAT et/ou ALAT > 5 N mais ≤ 20 N avec reprise à dose diminuée de 40 mg/jour si retour à une valeur < 3 N. Une interruption définitive du traitement doit, quant à elle, être proposée en cas de récidive, d’ASAT et/ou ALAT > 20 N ou d’ASAT et/ou ALAT > 3N avec bilirubine totale > 2 N. Concernant les autres effets indésirables, le tableau 4 résume la conduite à tenir vis-à-vis du traitement en fonction du grade de toxicité.
Les données des essais randomisés de phase III ont montré que la toxicité du régorafénib est maximale au cours des deux à trois premiers cycles de traitement et s’améliore ensuite [6, 13, 14]. Une surveillance attentive et rapprochée des patients au cours des deux premiers mois de traitement est ainsi recommandée, afin de détecter et de gérer efficacement les événements indésirables précoces. L’équipe du MD Anderson Cancer Center (Houston, Texas) a proposé une surveillance hebdomadaire, téléphonique ou en consultation, pendant les deux premiers mois de traitement [21]. Une surveillance biologique du bilan hépatique (hebdomadaire initialement puis tous les 15 jours) et de l’hémogramme (tous les 15 jours) est également recommandée. Les principaux effets secondaires du régorafénib sont le syndrome pied-main, qu’il faut tenter de prévenir, l’HTA et la diarrhée, surtout marqués en début de traitement. Une surveillance biologique rapprochée initiale est à prévoir
Trifluridine/tipiracil
Dans les deux études de phase III princeps européenne et asiatique, le taux de toxicité grade 3-4 du trifluridine/tipiracil a été de 45 % et 69 % [7, 9]. Le profil de toxicité du trifluridine/tipiracil est largement dominé par la toxicité hématologique, touchant préférentiellement les polynucléaires neutrophiles et l’hémoglobine, mais aussi à un moindre degré les plaquettes, avec des taux de neutropénie, anémie et thrombopénie de grade 3-4 autour de 35 %, 18 % et 5 % respectivement (tableau 2). Le taux de neutropénie fébrile était cependant assez faible, de seulement 4 % dans l’étude RECOURSE [7], et étonnamment aucun cas n’était rapporté dans l’étude asiatique TERRA [9]. Si les effets indésirables de grade 3-4 sont ceux qui attirent le plus l’attention du clinicien car ils vont systématiquement engendrer une adaptation de dose, il ne faut cependant pas négliger les toxicités hématologiques moins sévères, de grade 1-2, qui peuvent indéniablement avoir un impact sur la qualité de vie des patients (anémie responsable de fatigue, dyspnée…), mais aussi sur la prise en charge thérapeutique (report de cure en cas de thrombopénie persistante). La fréquence et l’importance de la neutropénie sous trifluridine/tipiracil pose la question de l’intérêt d’une prophylaxie primaire par G-CSF. Cependant, le faible taux de neutropénie fébrile n’est pas en faveur de cette attitude de manière systématique car largement sous le seuil de 20 % retenu par les recommandations américaines et européennes concernant l’utilisation des facteurs de croissance leucocytaires en cancérologie [22, 23].
Les autres effets indésirables les plus fréquents du trifluridine/tipiracil sont les effets digestifs (nausées, vomissements, diarrhée), observés dans 15 % à 50 % des cas, la perte d’appétit et la fatigue, ces effets indésirables non hématologiques étant le plus souvent de grade 1-2 (tableau 2). Des anomalies biologiques sont également possibles mais moins fréquemment rapportées (< 10 % des cas) : augmentation de la créatininémie, hypo ou hyperkaliémie, hypo- ou hypercalcémie, hyponatrémie, hypoalbuminémie. En pratique, après mise en route du trifluridine/tipiracil, une surveillance clinique mensuelle est recommandée, avec, si possible, appel téléphonique par l’infirmière de coordination à J15, ainsi qu’une surveillance de l’hémogramme et de l’ionogramme hebdomadaire le premier mois puis tous les 15 jours. Le tableau 5 résume les effets indésirables qui nécessitent une réduction de la posologie du trifluridine/tipiracil par palier de 5 mg/m2. La toxicité du trifluridine/tipiracil est surtout hématologique, touchant préférentiellement les polynucléaires neutrophiles et l’hémoglobine, et peut entraîner des adaptations posologiques
Biomarqueurs prédictifs
Différentes caractéristiques cliniques ou biologiques ont été analysées dans le but d’identifier des sous-groupes de patients ayant le meilleur bénéfice du régorafénib ou du trifluridine/tipiracil : origine géographique, âge, siège de la tumeur primitive, statut tumoral KRAS (exon 2), traitement antérieur par bévacizumab ou anti-EGFR, survenue d’effets indésirables spécifiques ou réponse radiologique particulière.
Origine géographique
Dans l’étude RECOURSE, la médiane de survie globale avec le trifluridine/tipiracil a été supérieure au Japon (7,8 mois) par rapport aux États-Unis (6,5 mois) et en Europe (6,8 mois). Cependant, en comparaison avec le bras placebo, le HR a été de 0,56 pour les États-Unis et de 0,64 pour l’Europe, et moindre pour le Japon (HR : 0,75). L’étude asiatique TERRA renforce ces résultats avec une médiane de survie globale de 7,8 mois dans le bras trifluridine/tipiracil (placebo : 7,1 mois) et un HR de 0,79 [9], suggérant donc un bénéfice supérieur du trifluridine/tipiracil chez les patients non asiatiques.
À l’inverse avec le régorafénib, la médiane de survie globale a été de 6,4 mois (placebo : 5,0 mois) dans l’étude internationale CORRECT (HR : 0,77) [6] et de 8,8 mois dans l’étude asiatique CONCUR (placebo : 6,3 mois ; HR : 0,55) [8], suggérant un bénéfice supérieur chez les patients asiatiques. Toutefois, l’effet de l’origine géographique reste spéculatif et sujet à caution. Par exemple, dans l’étude RECOURSE, les patients japonais avaient globalement une maladie moins symptomatique (PS 0 : 71 %) que les patients européens (PS 0 : 51 %) ou américains (PS 0 : 41 %) [24].
Âge
Dans l’étude RECOURSE, l’âge n’a pas été un facteur discriminant du bénéfice de survie globale du trifluridine/tipiracil tant chez les patients de plus de 65 ans (7,0 vs. 4,6 mois ; HR : 0,62) que chez ceux de plus de 70 ans (7,0 vs. 4,7 mois ; HR : 0,65) [24]. Dans l’étude rétrospective japonaise REGOTAS, la médiane de survie globale a été de 7,9 mois avec le régorafénib (223 patients) et de 7,4 mois avec le trifluridine/tipiracil (327 patients) [25]. Elle a été supérieure chez les patients de moins de 65 ans dans le groupe régorafénib (10,4 vs. 6,2 mois), mais non dans le groupe trifluridine/tipiracil (7,0 vs. 7,7 mois). Les auteurs concluent à une survie plus favorable avec le régorafénib pour les patients de moins de 65 ans.
Siège de la tumeur primitive
Dans l’étude observationnelle CORRELATE, la médiane de survie globale a été similaire avec le régorafénib chez les patients dont la tumeur primitive était localisée à gauche ou à droite (6,7 mois vs. 6,3 mois) [26]. Il n’y a donc pas d’argument pour penser que la localisation tumorale primitive ait un impact sur le bénéfice de ce traitement.
Statut mutationnel tumoral de KRAS
Dans l’étude RECOURSE, la médiane de survie globale a été de 8,0 mois avec le trifluridine/tipiracil dans la population avec une tumeur KRAS exon 2 sauvage contre 5,7 mois avec le placebo (HR : 0,58), et de 6,5 mois vs. 4,9 mois en cas de tumeur KRAS mutée (HR : 0,80) [24]. Dans l’étude CORRECT, la médiane de survie globale a été de 7,3 mois avec le régorafénib en cas de tumeur KRAS sauvage contre 5,0 mois avec le placebo (HR : 0,65), et de 6,2 mois vs. 5,1 mois en cas de tumeur KRAS mutée (HR : 0,87) [6]. Les auteurs concluent à une valeur prédictive discutable du statut tumoral KRAS (exon 2), les patients bénéficiant de chacun de ces deux traitements que leur tumeur soit KRAS exon 2 sauvage ou mutée.
Traitement antérieur par bévacizumab ou anti-EGFR
Dans l’étude CONCUR, à l’inverse de l’étude princeps CORRECT, certains patients du bras régorafénib n’avaient pas reçu de bévacizumab ou d’anti-EGFR avant participation à l’essai [8]. Le bénéfice de survie globale a été net chez les patients naïfs de ces traitements (9,7 mois vs. 4,9 mois ; HR : 0,31) et moindre chez ceux prétraités par une ou l’autre des deux classes d’anticorps (7,4 vs. 6,7 mois ; HR : 0,78).
Effets indésirables spécifiques
Une étude exploratoire de l’étude CORRECT a suggéré que le syndrome main-pied pouvait être un facteur prédictif de la survie globale, qu’il apparaisse au cycle 1 (7,2 vs. 5,7 mois ; HR : 0,66), au cycle 2 (8,0 mois vs. 5,3 mois ; HR : 0,57) ou quel que soit le cycle (9,5 vs. 4,7 mois ; HR : 0,41) [27]. Dans l’étude RECOURSE, un bénéfice net de survie globale du trifluridine/tipiracil était noté pour les patients avec neutropénie grade 3-4 au cycle 1 (9,7 mois vs. 5,3 mois ; HR : 0,53), au cycle 2 (8,7 mois vs. 6,3 mois ; HR : 0,56) et au-delà du cycle 2 (16,4 mois vs. 10,2 mois ; HR : 0,36) [30]. En l’absence de neutropénie grade 3-4, les médianes de survie globale étaient identiques entre le trifluridine/tipiracil et le placebo (5,5 mois vs. 5,3 mois ; HR : 0,97).
Réponses radiologiques particulières
L’évaluation rétrospective de 108 patients inclus dans l’étude CORRECT (75 dans le bras régorafénib et 33 dans le bras placebo) a permis de corréler l’absence de progression tumorale à 8 semaines avec la formation d’une cavitation dans les nodules métastatiques pulmonaires (29/75 avec le régorafénib vs. 0/33 avec le placebo ; p < 0,01) [28].
Au total, l’ensemble de ces données exploratoires ne permet pas d’individualiser clairement des facteurs prédictifs d’efficacité du régorafénib ou du trifluridine/tipiracil en traitement au-delà de la deuxième ligne du cancer colorectal métastatique. Le choix dépendra donc essentiellement des toxicités des traitements préalablement administrés, de la préférence du patient, et de l’appréciation clinique du prescripteur. Il peut être précisé que les patients traités dans l’étude CORRECT étaient naïfs de trifluridine/tipiracil [6]. À l’inverse, 144 patients de l’étude RECOURSE avaient initialement reçu du régorafénib [7]. Le bénéfice du trifluridine/tipiracil était similaire dans les deux sous-groupes avec un HR de 0,69. Très récemment, l’étude randomisée de phase II REVERCE a évalué chez des patients avec tumeur KRAS sauvage prétraités par fluoropyrimidine, oxaliplatine et irinotécan, la séquence régorafénib suivi de cétuximab (+ irinotécan) à progression (R-C) ou la séquence inverse (C-R) [29]. L’étude, par défaut d’inclusion, a été interrompue à 101 patients (180 initialement prévus). La survie globale (critère de jugement principal) a été en faveur du bras R-C (17,4 vs. 11,6 mois ; HR : 0,61 ; p = 0,029). Alors que la médiane de survie sans progression était de 2,4 mois pour la séquence R-C vs. 4,2 mois pour la séquence C-R (HR : 0,97 ; p = 0,91), la médiane de survie jusqu’à deuxième progression était de 5,2 mois pour la séquence R-C vs. 1,8 mois pour la séquence C-R (HR : 0,29 ; p < 0,0001). Les auteurs ont conclu à une supériorité de la séquence R-C. Cette étude est en faveur du concept de blocage continu de l’angiogenèse au-delà de la progression avec introduction des anticorps anti-EGFR le plus tard possible dans la séquence thérapeutique lorsqu’ils ne sont pas prescrits en première ligne. Quoiqu’il en soit, cette stratégie thérapeutique ne peut être proposée actuellement, l’AMM conditionnant l’utilisation du régorafénib aux patients prétraités par anticorps anti-EGFR en cas de tumeur RAS sauvage.
Enfin, on retiendra qu’actuellement il n’est pas possible de préconiser une séquence d’administration du régorafénib et du trifluridine/tipiracil plutôt qu’une autre en l’absence d’essai randomisé stratégique d’administration séquentielle de ces deux molécules, les recommandations internationales disponibles n’émettant aucune recommandation particulière à ce jour. Il n’existe pas de facteurs prédictifs de l’efficacité du régorafénib ou du trifluridine/tipiracil ni de données disponibles concernant la séquence thérapeutique optimale de ces médicaments au-delà de la deuxième ligne
Autres options thérapeutiques au-delà de la deuxième ligne
Elles sortent du sujet principal de cet article et ne seront donc que succinctement énumérées ici (tableau 6). Il importe de garder à l’esprit qu’à l’exception des anti-EGFR chez les patients avec tumeur RAS sauvage (lorsqu’ils n’ont pas été administrés au cours des deux premières lignes thérapeutiques), aucune de ces approches thérapeutiques ne peut être considérée comme un standard, en raison d’un niveau de preuve scientifique insuffisant (absence d’essais randomisés positifs). De fait, elles devraient toujours être discutées en réunion de concertation pluridisciplinaire, et leur rapport bénéfice/risque et leurs contraintes soigneusement pesées au regard des deux seuls standards validés dans cette situation : les deux monothérapies orales que constituent le régorafénib et le trifluridine/tipiracil.Take home messages
Liens d’intérêts
D. Malka : Amgen, Bayer, HalioDx, Merck Serono, MSD, Pierre Fabre Oncologie, Roche, Sanofi, Servier. A. Lièvre : Amgen, Bayer, BMS, Merck, Pierre Fabre Oncologie, Roche, Servier. R. Coriat : Merck, Amgen, Bayer, Roche. G. Lledo : HalioDx, Roche, Merck, Bayer, Pierre Fabre Oncologie. J. Bennouna : Astra Zeneca, Boehringer-Ingelheim, BMS, MSD, Servier, Roche.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International