Hépato-Gastro & Oncologie Digestive
MENUPARTIE 2 : Atteintes veineuses et sinusoïdales Volume 25, supplément 2, Septembre 2018

Syndrome de Budd-Chiari
P. Potier1, A. Coilly2 et P. Broué3
1 Service d’hépato-gastroentérologie, CHR Orléans
2 Centre hépatobiliaire, APHP-Hôpital Paul Brousse
3 Service d’hépatologie pédiatrique, CHU Toulouse
Relecteurs :
D. Valla4, S. Hillaire5, V. Lambert6, D. Dutheil7, C. Bureau8, A. Plessier4
4 Service d’hépatologie, APHP Hôpital Beaujon et centre de référence des maladies vasculaires du foie
5 Service de Gastro-entérologie hépatologie, Hôpital Foch, Paris
6 Médecin généraliste, Angers
7 Association des malades des vaisseaux du foie (AMVF)
8 Service d’hépato-gastroentérologie, CHU Toulouse
Introduction
Le syndrome de Budd-Chiari (SBC) est caractérisé par une obstruction du flux veineux hépatique dans l’espace vasculaire situé entre les veinules hépatiques et la jonction entre la veine cave inférieure et l’oreillette droite, excluant donc les causes d’amont (syndrome d’obstruction sinusoïdale) et d’aval (causes cardiaques) [1]. Dans les pays occidentaux, le SBC résulte le plus souvent d’une obstruction veineuse par thrombose révélant un état pro-thrombotique sous-jacent (cf. chapitre « Facteurs de risque des maladies vasculaires hépatiques »). Les causes secondaires de SBC par compression ou envahissement d’étiologie tumorale ou parasitaire sont exclues du champ de cette recommandation.
Épidémiologie adulte
L’épidémiologie du SBC n’est connue que de façon partielle, variable selon les régions du globe. Une enquête nationale française, menée en 2010 dans 48 centres de compétence du réseau national (maladies rares) des maladies vasculaires du foie, mesurait une incidence de 0,68 nouveaux cas et une prévalence de 4,04 cas par million d’habitants [2]. Une prédominance féminine (70 %) et un âge moyen de 38,6 ans caractérisaient la population étudiée. Une étude basée sur le programme de médicalisation des systèmes d’information (PMSI) enregistrait en 2012 une incidence de 2,17 par million d’habitants.
Épidémiologie pédiatrique
Le syndrome de Budd-Chiari est très rare chez l’enfant : moins de 0,1 % des hépatopathies chroniques de l’enfant dans les pays occidentaux, mais jusqu’à 16 % dans certains pays d’Asie [3]. Il peut s’observer à tout âge pédiatrique et peut être soupçonné devant une ascite fœtale.
Faire le diagnostic
Manifestations cliniques
Chez l’adulte
L’obstruction veineuse sus-hépatique induit, d’une part, une congestion sinusoïdale responsable d’une augmentation de volume du foie et, d’autre part, une ischémie et une nécrose hépatocytaire. Une hyperplasie nodulaire régénérative peut survenir, de même qu’une fibrose centrolobulaire susceptible d’évoluer vers une cirrhose. Le segment I du foie, de par son drainage veineux indépendant des veines hépatiques, est fréquemment le siège d’une hypertrophie compensatrice. Des lésions hépatocytaires bénignes ou malignes peuvent survenir au cours de l’évolution du SBC.
Le développement de réseaux veineux de dérivations collatérales limitant l’impact parenchymateux de la congestion veineuse explique l’existence de formes infra-cliniques. À l’opposé, le SBC peut se manifester de façon très aiguë, notamment en cas d’obstruction simultanée des trois veines hépatiques, par un tableau d’insuffisance hépatique aiguë associée à une encéphalopathie hépatique. Il s’agit d’une situation rare représentant moins de 1 % des hépatites aiguës graves mais qui engage le pronostic vital. La présence d’une hépatomégalie, inhabituelle au cours d’une hépatite aiguë grave, ainsi qu’un ratio aspartate aminotransférase/alanine aminotransférase supérieur à 1 pourraient orienter vers un SBC [4]. Entre ces deux présentations extrêmes, le SBC répond à une grande variété de tableaux cliniques, l’association d’une hépatomégalie et d’une ascite constituant les circonstances les plus fréquentes de diagnostic. La fréquence relative des symptômes révélateurs a été évaluée : ascite 83 %, hépatomégalie 67 %, douleurs abdominales 61 %, présence de varices œsophagiennes 58 %, hémorragie digestive 5 % [1]. La survenue d’un ictère ou d’une encéphalopathie hépatique semble moins fréquente. Une circulation collatérale thoracique sous-cutanée constitue un symptôme spécifique mais peu sensible de SBC par thrombose de la veine cave inférieure. Une thrombose porte associée est constatée dans 15 % des cas.
Chez l’enfant
La majorité des syndromes de Budd-Chiari de l’enfant se présente tardivement et reflète une évolution chronique : hépatomégalie ferme avec bilan hépatique normal ou peu perturbé jusqu’à un tableau de cirrhose avec hypertension portale, ascite, ictère, retard de croissance voire encéphalopathie. Plus rarement le tableau est aigu avec ascite, œdèmes, douleurs abdominales, hépatomégalie et progression rapide vers une insuffisance hépatique parfois fulminante [5].
Outils diagnostiques
Chez l’adulte
Le taux de protéines dans l’ascite varie mais une concentration de plus de 30 g/L est évocateur d’un SBC en dehors d’une cause cardiaque ou péricardique. La faible spécificité du tableau clinique et sa grande variabilité conduisent à recommander de suspecter un SBC dans toute situation de maladie du foie aiguë ou chronique, notamment lorsque la recherche des causes habituelles s’avère infructueuse.
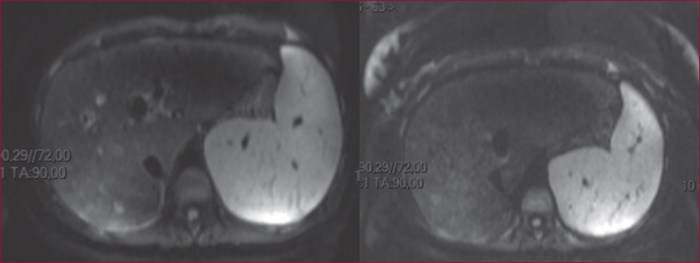
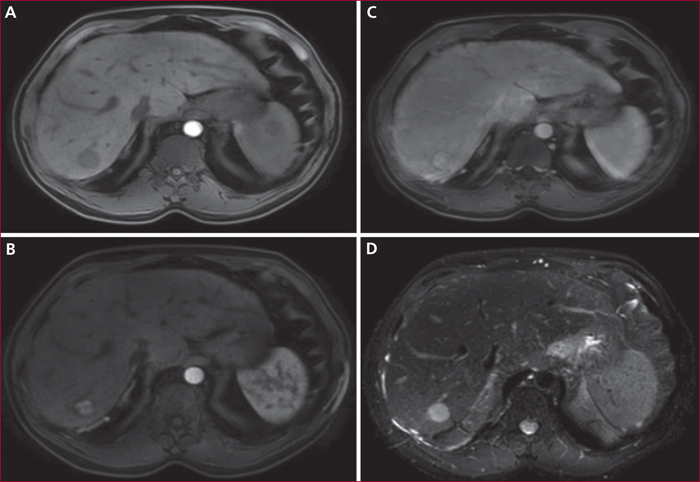
L’échographie-doppler hépatique constitue l’exploration de première intention en raison d’une sensibilité supérieure à 75 %, à la condition d’un opérateur expérimenté et sensibilisé à l’hypothèse d’un SBC [1]. Les critères diagnostiques considérés comme spécifiques comprennent l’absence ou des perturbations du flux sanguin dans une veine hépatique, la détection d’un réseau veineux collatéral intrahépatique ou sous-capsulaire. Une imagerie en coupe, notamment la tomodensitométrie (TDM) et l’imagerie par résonance magnétique (IRM), peut aussi montrer un aspect en toile d’araignée remplaçant l’image habituelle de veine hépatique au voisinage de l’ostium, une image hyperdense à l’emplacement habituel d’une veine hépatique, la survenue de troubles de la perfusion hépatique, aspect dit « en mosaïque » ou « en feuille de fougère » souvent attribué à la congestion et à la dilatation sinusoïdale, un rehaussement du segment I du foie lors de l’acquisition précoce après l’injection de produit de contraste. Elle permet également la recherche de nodules hépatiques (figures 1 et 2).
L’opacification percutanée directe des veines hépatiques est l’examen invasif historique de référence. Elle garde une place en cas de doute diagnostique et pour réaliser l’étude anatomique pré-thérapeutique immédiate. Les performances diagnostiques de l’IRM seraient cependant supérieures à celles de l’opacification veineuse pour la détection des veines collatérales. La biopsie hépatique n’est habituellement pas nécessaire lorsqu’une obstruction des gros troncs veineux a été diagnostiquée. En dehors de ce cas, elle peut permettre le diagnostic de SBC limité aux veinules hépatiques. Les résultats de l’élastométrie impulsionnelle sont modifiés par la congestion hépatique secondaire au SBC, limitant son utilisation pour évaluer le degré de fibrose hépatique. L’élastométrie pourrait cependant contribuer à apprécier l’évolution de cette congestion après traitement instrumental [6].
Chez l’enfant
Comme chez l’adulte, le diagnostic est le plus souvent établi par l’échographie mais nécessite un opérateur entraîné [3, 5, 7]. Les imageries en coupes permettent de compléter le diagnostic et de préciser les lésions : obstruction isolée des trois veines hépatiques (deux tiers des cas), ou combinée avec celle de la veine cave supérieure (un quart des cas). L’enquête étiologique est la même que chez l’adulte et doit rechercher, minutieusement, une maladie thrombogène [3, 5]. Les risques de complication tumorale sont identiques, y compris l’hépatocarcinome [8].
La prise en charge thérapeutique
Prise en charge thérapeutique chez l’adulte
Une fois le diagnostic de SBC établi, trois objectifs diagnostiques complémentaires peuvent être définis : identifier la situation de thrombophilie sous-jacente ou la maladie favorisante (Cf. le chapitre « Facteurs de risque des maladies vasculaires hépatiques »), établir le pronostic (fibrose avancée ou cirrhose, réponse au traitement, ascite réfractaire, dénutrition), caractériser d’éventuelles lésions hépatiques focales associées. Cette évaluation initiale contribue à déterminer le programme personnalisé de soin, fondé sur une stratégie par étape [9,10].
La décision de changer de stratégie est fondée sur la présence de trois critères qui définissent une réponse, réévaluée chaque semaine : (1) baisse de la bilirubine, (2) augmentation du facteur V, (3) disparition de l’ascite, y compris sous régime peu salé et faible dose de diurétiques. La diminution de la créatininémie et l’augmentation de la natrémie constituent également des critères de réponse au traitement.
Première étape : évaluer le pronostic
Si plusieurs scores prédictifs d’évolution ont été étudiés comme le score de Child-Pugh, le score MELD (Model for End Stage Liver Disease) ou des scores plus spécifiques comme les scores de Clichy, de Rotterdam, ou le score BCS-TIPS (Budd-Chiari Syndrome- Transjugular Intrahepatic Portosystemic Shunt), la prédiction individuelle du pronostic reste difficile. Le taux de survie spontanée à trois ans décrit comme défavorable dans les études anciennes (10 %) s’avère en nette amélioration dans les études plus récentes associées à la mise en œuvre des traitements recommandés, de l’ordre de 85 % à cinq ans [4]. Dans le cas de la grossesse, le pronostic maternel au cours de la grossesse apparaît excellent en cas de SBC bien contrôlé. Le pronostic fœtal est caractérisé par un risque accru d’accouchement prématuré qui surviendrait dans trois quarts des cas. Le risque malformatif associé aux anti-vitamines K justifie avant six semaines d’aménorrhée un relai précoce par héparine de bas poids moléculaire [1].
Deuxième étape : traitement de la maladie causale et anticoagulation
En premier lieu, un traitement de la maladie causale doit être entrepris sans délai. Les avantages d’un traitement précoce de la cause (syndrome myéloprolifératif sous-jacent, maladie de Behcet, etc.) ont été suggérés dans deux analyses de cohorte rétrospective françaises [11].
Un traitement anticoagulant doit être débuté sans délai après le diagnostic et poursuivi au long cours avec comme objectif de réduire l’extension de la thrombose mais aussi de prévenir la survenue d’un nouvel événement thromboembolique. L’anticoagulation doit être curative, basée sur une héparine de bas poids moléculaire en première intention, puis relayée par les anti-vitaminiques K (cf. le chapitre « Prise en charge de l’anticoagulation chez les patients adultes atteints d’une maladie chronique parenchymateuse ou vasculaire du foie »). Un taux élevé de complications hémorragiques en cas d’anticoagulation (de 17 % à 50 % des patients) a été rapporté dans des séries rétrospectives et une série prospective [12]. Pour éviter cette complication, il convient de prendre en charge rapidement les symptômes liés à l’hypertension portale (ascite, varices). En l’absence d’étude dédiée, les recommandations de la cirrhose sont appliquées [13]. Une prise en charge collaborative avec des spécialistes du traitement anticoagulant est souhaitée, en particulier lorsque des gestes invasifs dont la ponction d’ascite, sont envisagés.
Troisième étape : recanalisation
En l’absence de recanalisation spontanée ou sous anticoagulant, deux stratégies ont été étudiées : la thrombolyse chimique et l’angioplastie, parfois couplées. Même si de bons résultats de la thrombolyse, couplée à l’angioplastie, ont été publiés, particulièrement en cas de thrombose récente, la survenue d’événements indésirables graves voire mortels (hémorragie) fait que cette stratégie doit être évaluée au cas par cas [14]. L’angioplastie, plus ou moins couplée à la mise en place de prothèses, est à réserver aux sténoses partielles et localisées, présentes chez un tiers des patients. Le risque de récidive de la sténose peut être prévenu par la mise en place d’une prothèse mais peut compliquer la prise en charge ultérieure (TIPS, transplantation). Finalement, ces gestes de recanalisation sont efficaces chez moins de 10 % des patients occidentaux à long terme [10]. De biens meilleurs résultats ont été récemment rapportés dans une cohorte asiatique [15].
Quatrième étape : dérivation
Les techniques de dérivation comprennent le TIPS et les shunts chirurgicaux. Historiquement, le shunt chirurgical a été la première technique, par interposition d’un greffon veineux jugulaire ou de prothèse, reliant le système porte au système cave (mésentérico-cave, le plus souvent). Plusieurs shunts sont cependant possibles et la décision technique doit être remise à une équipe experte. Les shunts chirurgicaux n’ont cependant pas démontré de bénéfice en termes de survie dans plusieurs séries anciennes (avant 2000) [16]. Aujourd’hui, les shunts chirurgicaux sont supplantés par le TIPS, lorsqu’il est techniquement faisable. En effet, le TIPS a un taux de morbi-mortalité plus faible que la chirurgie. Une étude européenne rétrospective multicentrique, incluant 124 patients traités par TIPS, a montré une excellente survie sans transplantation à 1 et 5 ans (88 % et 78 %, respectivement) et confirmée par une étude prospective [10, 17]. Un TIPS couvert doit être préféré car il permet de réduire la récidive et limite le risque de thrombose. Il est souvent nécessaire de ré-intervenir sur le TIPS (42 % des patients dans une étude récente), en particulier en cas de SMP, le TIPS doit être mis en place dans un centre expert du TIPS et des maladies vasculaires du foie [18].
Cinquième étape : transplantation hépatique
La transplantation hépatique doit être réservée aux patients en échec des traitements précédents. Cependant, elle doit être discutée d’emblée en cas de présentation fulminante en particulier si l’évolution s’avère rapidement défavorable malgré le traitement anticoagulant [4]. Certains patients pourraient certainement tirer aussi bénéfice d’une transplantation plus précoce mais le niveau de preuve est faible et des études seraient à conduire pour établir les facteurs prédictifs. Le score TIPS-BCS a été développé pour prédire la survie sans transplantation à 1 an des patients ayant un TIPS et validé dans une cohorte indépendante [10, 17]. Il est défini comme suit : 0,08 × âge (années) + 0,16 × bilirubine (mg/dL) + 0,63 × INR (International Normalized Ratio). Un score ≤ 7 points a une très bonne valeur prédictive négative. La décision de transplantation repose sur l’appréciation de plusieurs éléments : (1) le pronostic de la maladie causale, (2) les chances d’amélioration sans recours à la transplantation, (3) la présence d’un carcinome hépatocellulaire. La survie globale est de 71 % à 5 ans et 68 % à 10 ans après transplantation, dans une série pourtant ancienne de l’ELTR (European Liver Transplant Registry) [19]. Une étude récente a suggéré que le pronostic vital à 10 ans d’une transplantation hépatique pour SBC n’était pas modifié par la présence ou l’absence d’un syndrome myélo-prolifératif (SMP) lors du diagnostic [20]. Le problème peut être la récidive de la maladie initiale après transplantation qui justifie de poursuivre le traitement de la maladie causale et une anticoagulation curative à vie (qui peut être discuté si l’état prothrombotique a été corrigé par la transplantation).
Prise en charge thérapeutique chez l’enfant
En raison du faible nombre de publications et de cas pédiatriques, il est difficile d’étayer une stratégie thérapeutique spécifique pour l’enfant. Ainsi la prise en charge du syndrome de Budd-Chiari de l’enfant s’oriente franchement vers celle proposée chez l’adulte [3, 7, 8]. Les techniques de radiologie interventionnelle (angioplastie, pose de stent, TIPS) sont possibles à tout âge pédiatrique par des opérateurs entraînés [5, 7, 8]. La petite taille des vaisseaux, la disponibilité de matériel adapté et la poursuite de la croissance vasculaire limitent la faisabilité, le succès et la pérennité du résultat dans le temps. Un syndrome hépato-pulmonaire peut survenir chez 20 % des patients [8]. Les traitements anticoagulants au long cours exposent à plus de complications chez l’enfant. Les dernières données suggèrent de tenter rapidement, si les délais le permettent, une anticoagulation, voire une thrombolyse puis une plastie vasculaire ou un TIPS après traitement symptomatique de l’ascite et de l’hypertension portale [3, 5, 7, 8]. En cas d’impossibilité, d’échec, de syndrome hépato-pulmonaire ou si le patient est en insuffisance hépatique avancée d’emblée, il faut réaliser rapidement une transplantation hépatique dans les pays occidentaux [3].
La surveillance chez l’adulte
Il n’existe pas d’étude permettant de définir avec précision le rythme de surveillance, une fois le diagnostic posé et le patient stabilisé. Cependant, le risque de dégénérescence devrait amener à une consultation tous les six mois, avec une imagerie hépatique. L’incidence cumulée à cinq ans du carcinome hépato-cellulaire (CHC) est de 4 % [1]. Le diagnostic peut être difficile compte tenu de la présence de nodules bénins dits de régénération chez plus de 60 % des patients. Les nodules bénins font habituellement moins de 4 cm, souvent multiples et disséminés dans le parenchyme hépatique, hypervasculaires de sémiologie non spécifique en imagerie en coupe. Selon les recommandations de l’EASL (European Association for the Study of the Liver), une biopsie hépatique devrait être proposée si la taille du nodule est supérieure à 3 cm, s’il est hétérogène ou se lave au temps portal, si sa taille se modifie ou devant une élévation du taux d’alpha-fœtoprotéine [1]. Une étude récente a montré l’intérêt de l’analyse par immunohistochimie de ces nodules. Tous les sous-types d’adénomes sont retrouvés, avec un risque accru de transformation maligne pour tous ces sous types [21]. Compte tenu de la coexistence possible de multiples nodules de nature variée, les critères de greffe pour carcinome hépatocellulaire, lorsque celui-ci est confirmé, ne sont pas définis dans le cadre du SBC. Les critères du score AFP (alpha-fœto-protéine) ne sont pas validés dans cette population. En attendant, une analyse multidisciplinaire par des anatomopathologistes, radiologues, chirurgiens et hépatologues experts est nécessaire, avec quelquefois la nécessité de faire appel à une exception au score de MELD.
Les recommandations chez l’adulte
- •Évoquer le diagnostic de SBC au cours de toute maladie aiguë ou chronique du foie (A).
- •Pour établir le diagnostic, faire une échographie-doppler hépatique par un opérateur averti et entraîné. Compléter l’évaluation par TDM et/ou IRM injectées (A). Ne pas faire de biopsie hépatique pour établir le diagnostic si le SBC est confirmé par les examens d’imagerie (C).
- •Débuter sans délai une anticoagulation curative, sauf contre-indication majeure (A). Surveiller le traitement anticoagulant avec l’aide des spécialistes de l’hémostase.
- •Contacter un centre de compétence du réseau des maladies vasculaires du foie pour définir les modalités de la prise en charge (B).
- •Appliquer les recommandations de la cirrhose à la prise en charge des complications de l’hypertension portale (C).
- •Dépister le CHC avec une échographie et une alpha-fœtoprotéine sérique tous les 6 mois. Caractériser les nodules par une IRM. Discuter la prise en charge en réunion de concertation pluridisciplinaire (RCP) de tumeurs hépatiques dans un centre de compétence (B).
- •Informer les patients de l’existence des associations de patients dès l’annonce du diagnostic.
Références
1. European Association for the Study of the Liver. Electronic address, e.e.e., EASL Clinical Practice Guidelines : Vascular diseases of the liver. J Hepatol 2016 ; 64 (1) : 179-202.
2. Ollivier-Hourmand I, Allaire M, Goutte N, Morello R, Chagneau-Derrode C, Goria O, et al. The epidemiology of Budd-Chiari syndrome in France. Dig Liver Dis 2018 Apr 12. pii : S1590-8658(18)30701-1.
3. Nobre S, Khanna R, Bab N, Kyrana E, Height S, Karani J, et al. Primary Budd-Chiari Syndrome in Children : King's College Hospital Experience. J Pediatr Gastroenterol Nutr 2017 ; 65 (1) : 93-96.
4. Parekh J, Matei VM, Canas-Coto A, Friedman D, Lee WM ; Acute Liver Failure Study Group. Budd-Chiari syndrome causing acute liver failure : A multicenter case series. Liver Transpl 2017 ; 23 (2) : 135-142.
5. Kathuria R, Srivastava A, Yachha SK, Poddar U, Baijal SS. Budd-Chiari syndrome in children: clinical features, percutaneous radiological intervention, and outcome. Eur J Gastroenterol Hepatol 2014 ; 26(9) : 1030-8.
6. Mukund A, Pargewar SS, Desai SN, Rajesh S, Sarin SK. Changes in Liver Congestion in Patients with Budd-Chiari Syndrome following Endovascular Interventions : Assessment with Transient Elastography. J Vasc Interv Radiol 2017 ; 28 (5) : 683-687.
7. Singh SK, Sen Sarma M, Yadav R, Kumar S, Prasad R, Yachha SK, et al. Prognostic scoring systems and outcome of endovascular radiological intervention of chronic Budd-Chiari Syndrome in children. Liver Int 2018 ; 38 (7) : 1308-1315..
8. Sharma VK, Ranade PR, Marar S, Nabi F, Nagral A. Long-term clinical outcome of Budd-Chiari syndrome in children after radiological intervention. Eur J Gastroenterol Hepatol 2016 ; 28 (5) : 567-75.
9. Plessier A, Sibert A, Consigny Y, Hakime A, Zappa M, Denninger MH, et al., Aiming at minimal invasiveness as a therapeutic strategy for Budd-Chiari syndrome. Hepatology 2006 ; 44 (5) : 1308-1316.
10. Seijo S, Plessier A, Hoekstra J, Dell’era A, Mandair D, Rifai K, et al. Good long-term outcome of Budd-Chiari syndrome with a step-wise management. Hepatology 2013 ; 57 (5) : 1962-1968.
11. Desbois AC, Rautou PE, Biard L, Belmatoug N, Wechsler B, Resche-Rigon M, et al. Behcet's disease in Budd-Chiari syndrome. Orphanet J Rare Dis 2014 ; 9 : 104.
12. Rautou PE, Douarin L, Denninger MH, Escolano S, Lebrec D, Moreau R, et al. Bleeding in patients with Budd-Chiari syndrome. J Hepatol 2011 ; 54 (1) : 56-63.
13. de Franchis R, Baveno VIF. Expanding consensus in portal hypertension : Report of the Baveno VI Consensus Workshop : Stratifying risk and individualizing care for portal hypertension. J Hepatol 2015 ; 63 (3) : 743-52.
14. Smalberg JH, Spaander MV, Jie KS, Pattynama PM, van Buuren HR, van den Berg B, et al. Risks and benefits of transcatheter thrombolytic therapy in patients with splanchnic venous thrombosis. Thromb Haemost 2008 ; 100 (6) : 1084-8.
15. Tripathi D, Sunderraj L, Vemala V, Mehrzad H, Zia Z, Mangat K, et al. Long-term outcomes following percutaneous hepatic vein recanalization for Budd-Chiari syndrome. Liver Int 2017 ; 37 (1) : 111-120.
16. Zeitoun G, Escolano S, Hadengue A, Azar N, El Younsi M, Mallet A, et al. Outcome of Budd-Chiari syndrome: A multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999. 30 (1) : 84-9.
17. Garcia-Pagán JC, Heydtmann M, Raffa S, Plessier A, Murad S, Fabris F, et al. TIPS for Budd-Chiari syndrome : long-term results and prognostics factors in 124 patients. Gastroenterology 2008 ; 135 (3) : 808-15.
18. Hayek G, Ronot M, Plessier A, Sibert A, Abdel-Rehim M, Zappa M, et al. Long-term Outcome and Analysis of Dysfunction of Transjugular Intrahepatic Portosystemic Shunt Placement in Chronic Primary Budd-Chiari Syndrome. Radiology 2017 ; 283 (1) : 280-292.
19. Mentha G, Giostra E, Majno PE, Bechstein WO, Neuhaus P, O’Grady J, et al. Liver transplantation for Budd-Chiari syndrome: A European study on 248 patients from 51 centres. J Hepatol 2006 ; 44 (3) : 520-8.
20. Potthoff A, Attia D, Pischke S, Mederacke I, Beutel G, Rifai K, et al. Long-term outcome of liver transplant patients with Budd-Chiari syndrome secondary to myeloproliferative neoplasms. Liver Int 2015 ; 35 (8) : 2042-9.
21. Sempoux C, Paradis V, Komuta M, Wee A, Calderaro J, Balabaud C, et al. Hepatocellular nodules expressing markers of hepatocellular adenomas in Budd-Chiari syndrome and other rare hepatic vascular disorders. J Hepatol 2015 ; 63 (5) : 1173-80.
Thrombose porte récente non cirrhotique
A. Heurgué1, A. Payancé2, D. Habes3, S. Franchi-Abella4
1 Service d’hépato-gastroentérologie, CHU Reims
2 Service d’hépatologie, APHP Hôpital Beaujon
3 Service d’hépatologie pédiatrique, APHP-Hôpital Bicêtre
4 Service radiologie pédiatrique, APHP-Hôpital Bicêtre
Relecteurs :
B. Condat5, D. Valla6, S. Hillaire7, D. Debray8, D. Dutheil9, C. Bureau10, A. Plessier6
5 Service d’hépatologie, Centre hospitalier de Polynésie française
6 Service d’hépatologie, APHP Hôpital Beaujon et centre de référence des maladies vasculaires du foie
7 Service de Gastro-entérologie hépatologie, Hôpital Foch, Paris
8 Service d’hépato-gastro-entérologie, APHP Hôpital Necker
9 Association des malades des vaisseaux du foie (AMVF)
10 Service d’hépato-gastroentérologie, CHU Toulouse
Introduction
La thrombose récente de la veine porte correspond à la survenue récente d’un thrombus dans la veine porte et/ou dans les branches portales droite ou gauche. Le thrombus peut s’étendre aux veines mésentériques ou à la veine splénique. L’occlusion peut être totale ou partielle, et survenir chez des patients ayant déjà une obstruction ancienne d’une partie du système veineux portal.
L’incidence de thrombose de la veine porte récente ou chronique chez l’adulte est évaluée à 0,7/100 000 habitants/an et la prévalence à 3/100 000 habitants en Europe [1, 2]. Chez l’enfant la thrombose porte est plus rare, le diagnostic est le plus souvent fait au stade chronique de cavernome porte (cf. le chapitre « Cavernome porte ou thrombose porte chronique »).
Ce chapitre traite de la thrombose récente de la veine porte survenant en l’absence de cirrhose et/ou de malignité.
Diagnostic et évaluation initiale
Causes de la thrombose récente de la veine porte
La recherche d’une cause est indispensable pour une prise en charge optimale du patient. La thrombose récente de la veine porte est souvent causée par une combinaison de facteurs de risque locaux et généraux. Une affection prothrombotique générale et un facteur local est mis en évidence chez environ 60 % et 30 % des patients respectivement. Plusieurs facteurs peuvent être mis en évidence chez un même patient mais dans un tiers des cas, aucune cause n’est identifiée [3, 4]. La recherche d’une thrombophilie doit être la règle et fait appel aux mêmes tests qu’en cas de thrombose ancienne découverte au stade de cavernome (voir ce chapitre). Idéalement, le bilan de thrombophilie doit être effectué avant le début des anticoagulants sans toutefois retarder la mise en place de ce traitement. La recherche d’une cause locale se fait par la relecture attentive du scanner initial qui a permis le diagnostic (recherche de signe d’appendicite, ou d’une pathologie intestinale, gastrique, biliaire ou pancréatique) et par la réalisation d’une coloscopie à distance de la thrombose aiguë. En l’absence de cause identifiée, une ponction biopsie hépatique a permis d’identifier une maladie porto-sinusoïdale chez 20 % des patients ayant eu une biopsie hépatique dans ce contexte, en particulier lorsque les tests hépatiques étaient modifiés ou en cas de dysmorphie hépatique initiale.
Il existe peu de données dans la littérature traitant de la thrombose récente de la veine porte chez l’enfant. Il s’agit essentiellement de cas isolés [5-7]. En période néonatale, la survenue d’une thrombose récente de la veine porte est favorisée par une omphalite, un cathéterisme veineux ombilical, un sepsis intra-abdominal, une deshydratation et exceptionnellement par une malformation vasculaire à type d’abouchement anormal de la veine ombilicale ou d’un retour veineux pulmonaire anormal. Chez le grand enfant, les principales causes sont la pyléphlébite, la colectomie (dans un contexte de maladie inflammatoire du tube digestif), la splénectomie, l’embolisation de la rate, la drépanocytose et le traitement par l’asparaginase. Même si des cas de thrombophilie associée à des thromboses récentes de la veine porte ont été rapportés aussi bien chez le nouveau-né que chez le grand enfant, le rôle des facteurs de thrombophilie dans la thrombose récente de la veine porte est probablement moins important chez l’enfant que chez l’adulte [8].
Manifestations cliniques
La plupart des patients atteints de thrombose récente de la veine portent présentent une douleur abdominale aiguë. Toutefois l’intensité des symptômes est très variable d’un patient à l’autre. De ce fait, le diagnostic peut être méconnu et établi seulement au stade de cavernome portal. Cette présentation semble plus fréquente chez l’enfant. Les anomalies des tests hépatiques sont habituellement modérées et transitoires. Un syndrome de réponse inflammatoire systémique est souvent présent en cas de thrombose récente de la veine porte mais une infection locale ou systémique est objectivée chez seulement 20 % de ces cas. Une ascite transitoire, souvent de faible abondance et visible en imagerie, est présente chez la moitié des patients [3]. En raison de la diffusion de l’imagerie non-invasive, le diagnostic de thrombose récente de la veine porte est maintenant établi dans la grande majorité des cas au stade aigu chez l’adulte [1].
Complications immédiates
L’infarctus mésentérique est la complication immédiate la plus sévère de la thrombose veineuse porto-mésentérique récente. Le taux de mortalité est élevé, autour de 60 % en l’absence de traitement anticoagulant. La résection étendue d’intestin grêle est parfois nécessaire avec un risque de syndrome de grêle court. Chez l’adulte, l’initiation précoce du traitement anticoagulant a été associée à une incidence très faible de l’infarctus mésentérique [3]. Les données manquent chez l’enfant.
Le diagnostic de l’infarctus mésentérique veineux est difficile car les manifestations cliniques, biologiques et radiologiques ne sont pas spécifiques. Des douleurs abdominales sévères, persistantes malgré un traitement anticoagulant à dose curative ; une défaillance d’un organe (choc, insuffisance rénale, acidose métabolique, taux élevé de lactates artériels) ; une ascite importante ou des rectorragies doivent faire évoquer le diagnostic d’infarctus mésentérique. Le diabète de type 2 a été identifié comme un facteur de risque d’infarctus mésentérique [9]. Une étude récente montre que le risque de nécrose intestinale augmente lorsque les facteurs suivants sont associés : anse intestinale dilatée, taux de lactates > 2 mmol/L ou d’une défaillance d’organe [10].
Diagnostic
L’échographie-doppler est l’examen à réaliser de première intention dans le contexte de douleurs abdominales. Il permet de détecter une absence de flux dans la veine porte, ou un thrombus hyperéchogène dans la lumière de la veine porte mais ce dernier peut ne pas être visible [9]. La qualité et l’interprétation de l’échographie-doppler dépendent de l’expertise de l’opérateur, ce qui en constitue les limites. L’obstruction aiguë de la veine porte ainsi que son extension doivent être confirmés en urgence par scanner et/ou imagerie par résonance magnétique (IRM) avec injection de produit de contraste avec acquisition d’images en phase portale [4]. Les images acquises lors de la phase artérielle tardive ne sont pas optimales pour le diagnostic de thrombose récente de la veine porte. En cas de flux ralenti dans la veine porte, un diagnostic de thrombose peut être porté de façon abusive en raison d’un défaut de remplissage de la veine. L’échographie-doppler et l’IRM ont une sensibilité plus faible que l’imagerie par scanner pour le diagnostic de thrombose récente de la veine porte.
Le scanner, à la phase portale, montre l’absence de rehaussement des veines thrombosées occupées par le thrombus. Seront évalués : l’extension du thrombus dans les veines du territoire splanchnique (veines mésentériques, veine splénique), un éventuel facteur local et des signes éventuels d’ischémie de l’intestin. Une occlusion distale de la veine mésentérique supérieure, un épaississement homogène ou hétérogène de la paroi intestinale, un hématome pariétal spontanément hyperdense, un rehaussement pariétal diminué, une pneumatose pariétale, une dilatation des anses digestives, des anomalies du mésentère (épaississement ou densification, aéromésentérie), une ascite abondante et la présence d’air dans la veine porte sont des signes d’ischémie de l’intestin plus fréquemment observés chez les patients nécessitant une résection intestinale [9]. Le scanner a également une meilleure sensibilité que l’échographie-doppler ou l’IRM pour rechercher une cause locale de thrombose récente de la veine porte (appendicite aiguë, diverticulite aiguë, cancer du côlon…).
La détermination précise de la date de survenue du thrombus n’est pas facile. Un thrombus récent peut être défini comme un thrombus se produisant dans le cadre d’une douleur abdominale aiguë et/ou d’un syndrome de réponse inflammatoire systémique. Un caillot spontanément hyperdense dans la lumière de la veine porte sur un scanner non injecté peut suggérer que le thrombus date de moins de 30 jours après l’apparition des symptômes, mais ce signe radiologique est rare même en cas de caillot très récent. L’absence de cavernome portal est aussi utile. Même si un cavernome de petite taille peut être identifié dès 15 à 30 jours après le début des douleurs abdominales un cavernome volumineux associé à des varices œsophagiennes de grande taille signe une thrombose portale ancienne [11]. Toutefois, le cavernome portal est absent en cas d’obstruction unilatérale d’une branche portale et la thrombose aiguë peut se superposer à un cavernome ancien.
Prise en charge thérapeutique
Objectifs du traitement
L’objectif initial du traitement de la thrombose récente de la veine porte est de limiter l’extension de la thrombose et d’obtenir une recanalisation des vaisseaux thrombosés pour prévenir l’ischémie veineuse mésentérique à court terme et à plus long terme, prévenir l’apparition d’une hypertension portale et de ses complications [4, 7]. Le traitement de la cause et le traitement anticoagulant doivent être instaurés le plus rapidement possible.
Prise en charge thérapeutique
Anticoagulation
La recanalisation spontanée est un événement rare chez l’adulte et le grand enfant [8]. Aussi, une anticoagulation à dose curative est recommandée chez les malades avec thrombose récente de la veine porte. Un traitement par héparine de bas poids moléculaire est proposé à dose curative avec une surveillance de l’activité anti-Xa chez les patients à risque (obésité, insuffisance rénale, grossesse). Une initiation plus précoce d’un traitement anticoagulant semble être associée à un taux de recanalisation plus élevé. Une recanalisation complète est observée dans près de 50 % des cas quand le traitement anticoagulant a été débuté moins de 24 h après le diagnostic radiologique [12, 13]. En effet, dans une étude prospective de 95 patients, l’initiation précoce d’une anticoagulation à dose curative permettait une recanalisation à un an de la veine porte chez 39 %, de la veine splénique chez 80 % et de la veine mésentérique supérieure chez 73 % des patients [3]. Il a également été montré que l’obstruction de la veine splénique et la présence d’ascite au moment du diagnostic sont des facteurs associés à l’absence de recanalisation de la veine porte chez ces malades [3, 13]. Après une à deux semaines de traitement, un relais par anticoagulants oraux (anti-vitamines K) avec une cible d’INR (International Normalized Ratio) compris entre 2 et 3 peut être fait. La durée recommandée de traitement est de 6 mois [12]. Le traitement sera poursuivi au long cours s’il existe un état prothrombotique sous-jacent avec un risque élevé de récidive de thrombose. Des manifestations initiales d’ischémie mésentérique ou la persistance d’une thrombose veineuse mésentérique constituent des arguments pour le maintien du traitement anticoagulant au long cours. Actuellement il n’existe pas de données concernant l’utilisation chez ces malades des nouveaux anticoagulants oraux. Un programme hospitalier de recherche clinique (PHRC) est en cours afin d’évaluer l’efficacité préventive du Xarelto® en termes de récidive de thrombose veineuse profonde chez des patients avec un antécédent de thrombose porte aiguë ou ayant une thrombose portale chronique mais sans facteur de risque élevé de récidive de thrombose. Une étude italienne suggère que l’incidence de la thrombopénie induite à l’héparine (TIH) est plus élevée chez les patients avec une thrombose de localisation portale comparativement aux thromboses d’autres localisations [14]. Cette incidence de TIH semble plus faible chez les patients traités par héparine de bas poids moléculaire [14].
Chez l’enfant, le traitement anticoagulant est extrapolé des recommandations établies chez l’adulte bien que cette approche ne soit pas optimale compte tenu des spécificités diagnostiques, physiopathologiques et thérapeutiques propres à l’enfant, en particulier chez le nouveau-né. Ces spécificités tiennent à un équilibre coagulolytique précaire propre à la période néonatale. L’utilisation des anticoagulants oraux (anti-vitamines K) est évitée avant l’âge de deux mois. La préférence est accordée à l’héparine de bas poids moléculaire qui est associée à un moindre risque hémorragique et de thrombopénie secondaire [15]. Certaines études chez le nouveau-né ont observé 70 % de recanalisation spontanée lorsque le thrombus est partiellement occlusif et 30 % lorsque le thrombus est complètement occlusif [16]. Le rôle du traitement anticoagulant reste assez controversé chez le nouveau-né et sa prescription relève d’une expertise médico-radiologique.
Radiologie interventionnelle et exploration chirurgicale
La thrombolyse pourrait être discutée chez les malades dont les symptômes persistent malgré l’instauration précoce des anticoagulants, ou en cas de thrombose porto mésentérique étendue avec ascite radiologique ayant peu de perspectives de reperméabilisation, ou malades non opérables et présentant des signes avant-coureurs d’un infarctus mésentérique. Cependant aucune recommandation ne peut être formulée car l’ensemble de la littérature ayant évalué cette technique représente moins d’une centaine de malades. Dans une étude rétrospective de 20 malades avec une thrombose récente porto-mésentérique, la réponse au traitement thrombolytique était bonne, quelle que soit la voie d’abord mais au prix d’un taux élevé de complications, en particulier des hémorragies sévères. Le plus souvent, il s’agissait d’accidents hémorragiques nécessitant une transfusion sanguine [17]. La voie transjugulaire pourrait être privilégiée pour aborder la veine porte du fait d’une possible diminution des risques de complications de la thrombolyse. Les données sont trop limitées pour évaluer cette approche [18].
Dans le contexte particulier des thromboses portales aiguës et mésentériques supérieures post-opératoires, l’angioplastie par ballon (avec ou sans insertion de stent) sans thrombolyse a récemment été rapportée pour avoir un faible taux de complication et une bonne efficacité pour la recanalisation [19]. Enfin, une exploration chirurgicale urgente doit être réservée aux malades présentant des signes de péritonite, d’infarctus intestinal ou de perforation, ou lorsque les facteurs suivants sont associés : anse intestinale dilatée, taux de lactates > 2 mmol/L, ou une défaillance d’organe.
Antibiothérapie
En cas de pyléphlébite septique (thrombose septique de la veine porte se constituant au contact d’un foyer infectieux et se présentant cliniquement par un syndrome infectieux sévère, et en particulier des frissons), un traitement antibiotique initialement à large spectre même en l’absence de bactériémie positive doit être instauré. En cas d’abcès hépatique qui peut assez souvent accompagner une pyléphlébite septique, il est proposé de maintenir l’antibiothérapie pendant six semaines [20].
Pronostic
Chez les patients sous anticoagulants, la recanalisation de la veine porte peut survenir jusqu’à 6 mois après la thrombose aiguë [3]. Cependant, dans la majorité des cas les vaisseaux non reperméabilisés à 3 mois resteront thrombosés par la suite. En cas de non-recanalisation, des signes d’hypertension portale (varices œsophagiennes, hypersplénisme) ou des complications tardives plus rares peuvent survenir comme une biliopathie portale secondaire à une compression par le cavernome porte [11], un syndrome hépato-pulmonaire, une hypertension porto-pulmonaire, une entéropathie exsudative. La survie à 5 ans de ces malades est généralement bonne, supérieure à 70 %. Les données de survie manquent chez l’enfant mais le pronostic à court terme semble plus favorable que chez l’adulte.
Recommandations
- •Évoquer le diagnostic de thrombose de la veine porte (TVP) chez tout patient présentant des douleurs abdominales (B1).
- •Dépister une thrombose de la veine porte chez les nouveau-nés à risque : sepsis sur cathétérisme de la veine ombilicale, omphalite et en cas de diagnostic anténatal de malformation vasculaire hépatique.
- •Faire une échographie-doppler en première intention pour le diagnostic de thrombose récente de la veine porte. Confirmer le diagnostic de TVP récente par un scanner en urgence, évaluer l’étendue de la TVP et rechercher une cause locale et des signes d’infarctus intestinal (A1).
- •Éliminer une cirrhose, un envahissement tumoral.
- •Chercher une maladie vasculaire porto-sinusoïdale (C1).
- •Chercher les facteurs de risque de thrombose splanchnique (cf. chapitre Facteurs de risque des maladies vasculaires du foie chez l’adulte et l’enfant).
- •Suspecter un infarctus intestinal, en cas d’aggravation de l’état clinique, de douleurs abdominales sévères et persistantes, de rectorragies, d’ascite modérée à sévère ou de dysfonctionnement d’organe multiple et lorsque les facteurs suivants sont associés : anse intestinale dilatée, taux de lactates > 2 mmol/L, ou une défaillance d’organe (B1).
- •Faire une laparotomie exploratrice pour résection éventuelle d’intestin nécrosé en cas d’infarctus mésentérique (B1).
- •Initier un traitement anticoagulant dès confirmation du diagnostic de thrombose portale récente (A1). Les héparines de bas poids moléculaires doivent être préférées.
- •Chez l’adulte, poursuivre le traitement anticoagulant 6 mois (A1).
- •Chez l’enfant, la durée du traitement ne fait pas l’objet d’un consensus. Chez le nouveau-né, il est habituel de prescrire un traitement de 6 semaines à 3 mois selon les données de la surveillance échographique.
- •L’anticoagulation au long cours est recommandée en cas d’antécédent d’infarctus intestinal, d’antécédents personnels ou familial au premier degré de maladie thromboembolique, d’état pro-thrombotique fort ou de thrombose récidivante (B1). Pour les autres situations, le dossier doit être discuté de façon multidisciplinaire avec un centre expert et en particulier chez l’enfant.
- •Proposer un suivi dans une clinique des anticoagulants ou avec une équipe d’hémostase, lorsque c’est possible. Rechercher, et prendre en charge les autres facteurs de risque cardiovasculaires associés.
- •L’inclusion dans un essai thérapeutique multicentrique doit être envisagée (B1).
- •Informer les patients de l’existence des associations de patients dès l’annonce du diagnostic.
Références
1. Ageno W, Dentali F, Pomero F, Fenoglio L, Squizzato A, Pagani G, et al. Incidence rates and case fatality rates of portal vein thrombosis and Budd-Chiari Syndrome. Thromb Haemost 2017 ; 117 (4) : 794-800.
2. Rajani R, Bjornsson E, Bergquist A, Danielsson A, Gustavsson A, Grip O, et al. The epidemiology and clinical features of portal vein thrombosis : a multicentre study. Aliment Pharmacol Ther 2010 ; 32 : 1154-1162.
3. Plessier A, Darwish MS, Hernandez-Guerra M, Consigny Y, Fabris F, Trebicka J, et al. Acute portal vein thrombosis unrelated to cirrhosis : a prospective multicenter follow-up study. Hepatology 2010 ; 51 : 210-218.
4. European Association for the Study of the Liver. EASL Clinical Practice Guidelines : Vascular diseases of the liver. J Hepatol 2016 ; 64 (1) : 179-202.
5. Fraser CJ, Newell F, Furmedge J, Campbell J, Savoia H, Monagle PT. Acute idiopthi Portal vein Thrombosis in a child : case report and literature review. Thrombosis Research 2006 ; 117 : 279-281.
6. Degano LA, El Kik SA, Rizzi A. Pylephlebitis in pediatric patients. Arch Argent Pediatr 2014 : 112 (4) ; 163-166.
7. Di Francisco F, Del Prete L, Grimaldi C, Monti L, De Ville de Goyet J. Pancreatitis and portal vein Thrombosis in Children : The Chicken or the Egg Causality Dilemma. J Pediatric Surg 2015 ; 50 : 565-569.
8. Petrobattista A, Luciani M, Abraldes JG, candusso M, Pancotti S, Soldati M et al. Extrahepatic portal vein thrombosis in children and adolescents : influence of genetic thrmbophilic disorders. World J Gastroenterol 2010 ; 16 (48) : 6123-6127.
9. Elkrief L, Corcos O, Bruno O, Larroque B, Rautou PE, Zekrini K, et al. Type 2 diabetes mellitus as a risk factor for intestinal resection in patients with superior mesenteric vein thrombosis. Liver Int 2014 ; 34 (9) : 1314-1321.
10. Nuzzo A, Maggiori L, Ronot M, Becq A, Plessier A, Gault N, Joly F, Castier Y, Vilgrain V, Paugam C, Panis Y, Bouhnik Y, Cazals-Hatem D, Corcos O. Predictive Factors of Intestinal Necrosis in Acute Mesenteric Ischemia : Prospective Study from an Intestinal Stroke Center. Am J Gastroenterol 2017 ; 112 (4) : 597-605.
11. Plessier A, Rautou PE, Valla DC. Management of hepatic vascular diseases. J Hepatol 2012 ; 56 : S25-S38.
12. Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis : increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000 ; 32 : 466-470.
13. Turnes J, Garcia-Pagan JC, Gonzalez M, Aracil C, Calleja JL, Ripoll C, et al. Portal hypertension-related complications after acute portal vein thrombosis : impact of early anticoagulation. Clin Gastroenterol Hepatol 2008 ; 6 : 1412-1417.
14. Randi ML, Tezza F, Scapin M, Duner E, Scarparo P, Scandellari R, Fabris F. Heparin-induced thrombocytopenia in patients with Philadelphia-negative myeloproliferative disorders and unusual splanchnic or cerebral vein thrombosis. Acta Haematol 2010 ; 123 (3) : 140-5.
15. Hepponstall M, Chan A, Monagle P. Anticoagulation therapy in neonates, children and adolescents. Blood Cells, Molecules and Diseases 2017 ; 67 : 41-47.
16. Kim JH, Lee YS, Kim SH, Lee SK, Lim MK, Kim HS. Does umbilical vein catheterization lead to portal venous thrombosis? Prospective US evaluation in 100 neonates. Radiology 2001 ; 219 : 645-650.
17. Hmoud B, Singal AK, Kamath PS. Mesenteric venous thrombosis. J Clin Exp Hepatol 2014 ; 4 (3) : 257-63.
18. Wang MQ, Liu FY, Duan F, Wang ZJ, Song P, Fan QS. Acute symptomatic mesenteric venous thrombosis : treatment by catheter-directed thrombolysis with transjugular intrahepatic route. Abdom Imaging 2011 ; 36 (4) : 390-8.
19. Cao G, Ko GY, Sung KB, Yoon HK, Gwon DI, Kim JH. Treatment of postoperative main portal vein and superior mesenteric vein thrombosis with balloon angioplasty and/or stent placement. Acta Radiol 2013 ; 54 (5) : 526-32.
20. Choudhry AJ, Baghdadi YMK, Amr MA, Alzghari MJ, Jenkins DH, Zielinski MD. Pylephlebitis : A Review of 95 Cases. J Gastrointest Surg 2016 ; 20 (3) : 656-661.
Cavernome porte ou thrombose porte chronique
L. Elkrief1, P. Houssel-Debry2, O. Ackermann3, S. Franchi-Abella4, S. Branchereau5
1 Service d’hépato-gastroentérologie et de transplantation, Hôpitaux Universitaires de Genève (Suisse)
2 Service des maladies du foie, CHU Rennes
3 Service d’hépatologie pédiatrique, APHP Hôpital Bicêtre
4 Service de radiologie pédiatrique, APHP Hôpital Bicêtre
5 Service de chirurgie pédiatrique à orientation viscérale, APHP-Hôpital Bicêtre
Relecteurs :
D. Valla6, S. Hillaire7, D. Dutheil8, C. Bureau9, A. Plessier6
6 Service d’hépatologie, APHP Hôpital Beaujon et centre de référence des maladies vasculaires du foie
7 Service de gastro-entérologie hépatologie, Hôpital Foch, Paris
8 Association des malades des vaisseaux du foie (AMVF)
9 Service d’hépato-gastroentérologie, CHU Toulouse
Introduction
Après une thrombose aiguë de la veine porte (obstruction de la veine porte par un caillot) et en l’absence de recanalisation, un réseau de veines collatérales porto-porte, appelé « cavernome », se développe. Le cavernome est considéré comme la séquelle de la thrombose. Le terme cavernome est synonyme de thrombose chronique de la veine porte. Chez l’enfant, il existe des formes dans un contexte malformatif, après cathéter veineux ombilical ou omphalite. Environ 50 % restent idiopathiques [1].
Diagnostic et évaluation initiale
Manifestations
Chez l’adulte
Le diagnostic est fait au cours du suivi d’une thrombose porte aiguë ou, dans 30 % des cas, de manière fortuite par un examen d’imagerie effectué pour une autre raison [2].
Les manifestations liées à l’hypertension portale (HTP) sont les plus fréquentes. Les varices œsophagiennes (VO) sont présentes chez un patient sur deux [2]. Les autres signes comprennent des varices rectales, une splénomégalie. La fonction hépatique reste préservée ce qui contraste avec des manifestations évidentes d’hypertension portale. Une bicytopénie est fréquente liée à l’hypersplénisme.
La cholangiopathie portale est caractérisée par des anomalies des voies biliaires intra- et extrahépatiques, en l’absence d’autre cause de cholangiopathie. Elle correspond à la compression extrinsèque des voies biliaires intra- ou extrahépatiques par les veines du cavernome et/ou une ischémie des voies biliaires par thrombose de veinules dédiées à l’arbre biliaire [3]. Des anomalies des voies biliaires sont présentes chez 77 % à 100 % des malades avec un cavernome [4, 5]. Le plus souvent, la cholangiopathie portale est asymptomatique ou se manifeste par des anomalies isolées des enzymes hépatiques. Les manifestations biliaires sévères (colique hépatique, cholécystite, ictère obstructif, angiocholite, pancréatite) sont rares, survenant dans seulement 5 à 30 % des cas [4, 5].
Chez l’enfant
Les modes de révélation principaux sont la thrombopénie, la splénomégalie, les hémorragies inaugurales.
Les complications
Les complications les plus fréquentes sont l’hémorragie digestive liée à l’HTP, la récidive d’une thrombose (préférentiellement dans le territoire splanchnique) et les complications biliaires.
Comme dans la cirrhose, le risque d’hémorragie est surtout présent en cas de VO de moyenne ou grande taille [6].
La récidive de thrombose est souvent asymptomatique, donc sous-estimée et de conséquence mal évaluée. Ceci n’est pas observé chez l’enfant.
L’ascite, l’encéphalopathie hépatique, les infections bactériennes sont rares et le plus souvent transitoires. Elles surviennent préférentiellement au décours d’une hémorragie digestive [7]. La fréquence de l’encéphalopathie hépatique minime serait néanmoins de 35 % [8], non évaluée chez l’enfant.
Des nodules hépatiques de type « HNF-like » sont fréquemment retrouvés à l’IRM chez l’adulte. En revanche, le risque de carcinome hépatocellulaire ne semble pas augmenter chez les malades atteints de cavernome [6].
Les complications cardiovasculaires à type de shunts intra-pulmonaires et d’hypertension artérielle pulmonaire ont été rapportées [1].
Diagnostic
L’échographie-doppler hépatique, la tomodensitométrie et l’imagerie par résonance magnétique (IRM) avec injection de produit contraste vasculaire permettent de poser le diagnostic. Le diagnostic repose sur l’absence de flux dans la veine porte et la visualisation d’un réseau veineux collatéral correspondant au cavernome. Une dysmorphie hépatique qui associe une hypertrophie des segments I et IV et une atrophie du secteur latéral gauche est un signe indirect [9]. La découverte d’un cavernome en échographie-doppler doit faire réaliser un examen en coupes. En effet, la tomodensitométrie et l’IRM avec injection de produit de contraste vasculaire permettent d’apprécier l’extension de la thrombose et les signes d’HTP.
La cholangio-IRM est l’examen de référence pour le diagnostic de cholangiopathie d’HTP [3].
Une biopsie hépatique ne doit être envisagée que si une maladie hépatique chronique est suspectée (en particulier une cirrhose ou une veinopathie portale oblitérante) Une élasticité hépatique inférieure à 10 kPa permet d’exclure une cirrhose chez l’adulte [10].
Prise en charge thérapeutique
Prévention de l’extension et de la récidive de TVP
Les recommandations sur l’intérêt du traitement anticoagulant au long cours chez les malades atteints de thrombose de la veine porte (TVP) chronique reposent sur des séries rétrospectives chez l’adulte [11-14]. Les anticoagulants ont été associés à une diminution du risque d’extension ou de récidive de thrombose dans trois études [12-14], et à une amélioration de la survie dans une [15]. Les facteurs associés à la récidive de la thrombose était la présence d’un état pro-thrombotique [5, 13, 14]. Enfin, chez les malades avec un antécédent d’infarctus intestinal, le traitement anticoagulant au long cours était associé à une diminution du risque de récidive de thrombose [13]. Chez l’enfant, aucun traitement anticoagulant au long cours n’est institué.
Le traitement anticoagulant n’était associé à un risque augmenté d’hémorragie digestive par rupture de varices que dans une étude [14]. Les anticoagulants n’étaient jamais associés à la sévérité des hémorragies digestives.
Les anticoagulants les plus fréquemment utilisés sont les héparines (héparines non fractionnées (HNF) ou héparines de bas poids moléculaires (HBPM)) et les anti-vitamines K (AVK). Chez les malades atteints de TVP associée à un syndrome myélo-prolifératif (SMP) qui avaient reçu de l’HNF, la fréquence de la thrombopénie induite par l’héparine était accrue, allant jusqu’à 20 % [16]. L’utilisation des anticoagulants oraux directs (rivaroxaban, apixaban et dabigatran) a été très peu évaluée : une série descriptive rétrospective de moins de 30 malades atteints de TVP non cirrhotique ne rapporte pas de surrisque hémorragique [17], mais ces résultats doivent être confirmés dans une étude contrôlée.
Prise en charge des complications de l’hypertension portale
Chez l’adulte
Dans une étude récente ayant inclus 178 malades avec TVP, l’histoire naturelle des varices œsophagiennes semblait similaire à celle observée chez les malades atteints de cirrhose [2]. Chez les malades sans VO initialement, le risque d’en développer était de 2 % à 1 an, et 22 % à 5 ans. Chez les malades avec de petites VO initialement, le risque de développer des VO moyennes ou grosses était de 13 % à 1 an et 54 % à 5 ans. Chez les patients avec des VO moyennes ou grosses, et qui recevaient une prophylaxie, le risque de saignement était de 9 % à 1 an, et 32 % à 5 ans. Les bêta-bloquants non cardiosélectifs étaient associés à une diminution du risque hémorragique dans une étude [12] et à une amélioration de la survie dans une autre [15]. La fréquence des hémorragies était similaire chez les malades traités par bêta-bloquants ou par ligatures (32 % et 25 % respectivement) [2]. Dans une étude randomisée, l’efficacité des bêta-bloquants et de la ligature des VO était similaire pour prévenir la récidive hémorragique, chez des malades ne recevant pas d’anticoagulants [18]. Enfin, une étude évaluant 471 séances de ligature endoscopique, rapporte que la ligature de varices œsophagiennes (LVO) peut être effectuée sans surrisque hémorragique chez les patients traités par anticoagulant au cours du cavernome [19].
La pose d’un TIPS (Transjugular Intrahepatic Portosystemic Shunt) chez les malades avec cavernome est possible sous certaines conditions mais la place du TIPS reste à préciser [20]. La recanalisation portale par voie radiologique associée ou non à un TIPS peut être discutée lorsque le traitement médical et endoscopique ne permet pas de traiter les complications symptomatiques de l’HTP [21].
Chez l’enfant
Chez l’enfant aucune étude n’encourage l’utilisation des bêta-bloquants dans la prévention de l’hémorragie digestive [1]. En revanche, l’efficacité de la prophylaxie primaire endoscopique a été montrée [22].
Prise en charge de la cholangiopathie portale
Les recommandations sur le traitement de la cholangiopathie portale reposent sur des avis d’experts. Chez l’adulte, un traitement spécifique ne devrait être envisagé qu’en cas d’ictère, prurit, ou d’angiocholite [3]. Un traitement endoscopique est envisageable en cas de calculs de la voie biliaire principale ou de sténose biliaire. Le risque de saignement, lié à la présence de varices endobiliaires, doit être pris en compte. L’administration d’acide ursodéoxycholique après traitement endoscopique était associée à une absence de récidive des symptômes dans 50 % des cas [4, 5, 23]. Aucune n’étude n’a comparé le traitement par acide ursodéoxicholique au traitement endoscopique. Un shunt chirurgical porto-systémique de décompression peut également être envisagé [24]. Le pronostic des malades avec une cholangiopathie portale est excellent après traitement endoscopique et/ou décompression portale [3]. Les anastomoses bilio-digestives ne sont pas recommandées en première intention, en raison du risque de complications sévères dans 30 % des cas (angiocholite et hémorragie peropératoire) et de récidive des symptômes dans 70 % des cas [25].
Chez l’enfant, la cholangiopathie portale survient dans environ 6 % des cas. Certains enfants restent asymptomatiques mais développent des anomalies radiologiques et/ou de la biologie hépatique. L’évolution vers la fibrose hépatique est quasi systématique et des cas de cirrhose biliaire secondaire sont décrits (1 cas sur 8 dans la série de Bicêtre). La survenue d’une cholangiopathie portale doit donc faire discuter un traitement chirurgical de l’HTP en raison du risque de cirrhose biliaire secondaire [26]
Traitement chirurgical
Les recommandations concernant les indications du traitement chirurgical reposent sur des avis d’experts.
Chez l’adulte
Le shunt porto-cave reste une possibilité dans les rares cas de complications réfractaires de l’hypertension portale ou chez les patients présentant une cholangiopathie portale sévère. L’expérience de l’anastomose méso-Rex chez l’adulte est limitée à quelques cas cliniques.
Chez l’enfant
La reperfusion portale par anastomose méso-Rex, quand elle est envisageable, est indiquée chez l’enfant dans la prophylaxie primaire et secondaire de l’hémorragie digestive, en cas de cholangiopathie portale ou de complication cardio-pulmonaire [27]. La reperfusion portale peut être envisagée avec un bon taux de succès (93 %) quand le recessus de Rex est perméable et en l’absence d’extension de la thrombose au réseau splénique et mésentérique [28]. Elle est le traitement physiologique de référence. La reperfusion portale est contre-indiquée en cas de non perméabilité du réseau porte gauche et en cas de suspicion de veinopathie portale oblitérante associée au cavernome En l’absence de visualisation du système porte intrahépatique sur l’imagerie standard, la réalisation d’une phlébographie sus-hépatique bloquée est recommandée pour mettre en évidence des branches portes intrahépatiques hypoplasiques permettant la reperfusion portale [28].
La dérivation porto-systémique est indiquée en cas de persistance de complications hémorragiques malgré le traitement endoscopique, ou en cas de cholangiopathie symptomatique [29]. La dérivation n’est pas indiquée en cas de complications cardio-pulmonaires ou d’encéphalopathie hépatique. Dans ce dernier cas, la transplantation hépatique peut être envisagée [29]. Il existe plusieurs types de dérivations porto-systémiques chirurgicales, totales ou plus ou moins partielles [29]. Selon les études, on retrouve entre 53 % et 100 % de succès de ce type de chirurgie avec le risque de développer des complications cardio-pulmonaires, une encéphalopathie hépatique ou des nodules hépatiques [30, 31].
Recommandations
- •Évoquer le diagnostic de cavernome chez un patient présentant des signes d’hypertension portale ou des symptômes biliaires (A1).
- •Faire le diagnostic par la réalisation d’au moins une imagerie en coupe injectée 4 temps (A1).
- •Faire une biopsie hépatique en cas de suspicion de maladie hépatique associée ou avant reperfusion portale chez l’enfant (B1).
- •Traiter par anticoagulation au long cours en cas d’antécédent d’infarctus intestinal, d’antécédents personnels ou familial au premier degré de maladie thromboembolique, d’état pro-thrombotique fort (B1). Dans les autres cas, les données disponibles ne permettent pas de faire de recommandations pour ou contre le traitement anticoagulant : la discussion du dossier peut être soumise à un centre de compétence pour concertation multidisciplinaire (C).
- •La cholangio-IRM est l’examen de référence pour le diagnostic de cholangiopathie portale (B1).
- •Effectuer une endoscopie de dépistage des varices œsogastriques chez tous les patients atteints de cavernome (B1). Ne pas faire de relai systématique chez les patients sous anticoagulants, pour le traitement endoscopique. Il n’y a pas de sur-risque hémorragique de la LVO sous AVK, lorsque l’INR (International Normalized Ratio) est dans les valeurs cibles.
- •Chez l’adulte, appliquer les recommandations de la cirrhose à la prise en charge des complications de l’hypertension portale.
- •Chez l’enfant, les bêta-bloquants ne sont pas recommandés (C2). Le traitement endoscopique est recommandé en prophylaxie secondaire et a montré son efficacité en prophylaxie primaire (B2).
- •La reperfusion portale, quand elle est possible, est indiquée en cas de manifestations d’hypertension portale (B1).
- •Le traitement endoscopique ou chirurgical de la cholangiopathie portale ne doit être envisagé qu’en cas d’obstruction biliaire symptomatique (B1).
- •Informer les patients de l’existence des associations de patients dès l’annonce du diagnostic.
Références
1. Flores-Calderón J, Morán-Villota S, Solange-Heller R, Nares-Cisneros J, Zárate-Mondragón F, González-Ortiz B, et al. Guidelines for the diagnosis and treatment of extrahepatic portal vein obstruction (EHPVO) in children. Ann Hepatol 2013 ; 12 : 3-24.
2. Noronha Ferreira C, Seijo S, Plessier A, Silva-Junior G, Turon F, Rautou P-E, et al. Natural history and management of esophagogastric varices in chronic noncirrhotic, nontumoral portal vein thrombosis. Hepatol Baltim Md 2016 ; 63 : 1640-1650.
3. Dhiman RK, Saraswat VA, Valla DC, Chawla Y, Behera A, Varma V, et al. Portal cavernoma cholangiopathy : consensus statement of a working party of the Indian national association for study of the liver. J Clin Exp Hepatol 2014 ; 4 : S2-S14.
4. Llop E, Juan C de, Seijo S, García-Criado Á, Abraldes JG, Bosch J, et al. Portal cholangiopathy : radiological classification and natural history. Gut 2011 ; 60 : 853-860.
5. Condat B, Vilgrain V, Asselah T, O’Toole D, Rufat P, Zappa M, et al. Portal cavernoma-associated cholangiopathy: A clinical and MR cholangiography coupled with MR portography imaging study. Hepatol Baltim Md 2003 ; 37 : 1302-1308.
6. Marin D, Galluzzo A, Plessier A, Brancatelli G, Valla D, Vilgrain V. Focal nodular hyperplasia-like lesions in patients with cavernous transformation of the portal vein : prevalence, MR findings and natural history. Eur Radiol 2011 ; 21 : 2074-2082.
7. Rangari M, Gupta R, Jain M, Malhotra V, Sarin SK. Hepatic dysfunction in patients with extrahepatic portal venous obstruction. Liver Int 2003 ; 23 : 434-439.
8. Sharma P, Sharma BC, Puri V, Sarin SK. Minimal hepatic encephalopathy in patients with extrahepatic portal vein obstruction. Am J Gastroenterol 2008 ; 103 : 1406-1412.
9. Vilgrain V, Condat B, Bureau C, Hakimé A, Plessier A, Cazals-Hatem D, et al. Atrophy-hypertrophy complex in patients with cavernous transformation of the portal vein : CT evaluation. Radiology 2006 ; 241 : 149-155.
10. Sharma P, Mishra SR, Kumar M, Sharma BC, Sarin SK. Liver and spleen stiffness in patients with extrahepatic portal vein obstruction. Radiology 2012 ; 263 : 893-899.
11. Rajani R, Björnsson E, Bergquist A, Danielsson A, Gustavsson A, Grip O, et al. The epidemiology and clinical features of portal vein thrombosis : a multicentre study. Aliment Pharmacol Ther 2010 ; 32 : 1154-1162.
12. Condat B, Pessione F, Hillaire S, Denninger MH, Guillin MC, Poliquin M, et al. Current outcome of portal vein thrombosis in adults : risk and benefit of anticoagulant therapy. Gastroenterology. 2001 ; 120 : 490-497.
13. Amitrano L, Guardascione MA, Scaglione M, Pezzullo L, Sangiuliano N, Armellino MF, et al. Prognostic factors in noncirrhotic patients with splanchnic vein thromboses. Am J Gastroenterol 2007 ; 102 : 2464-2470.
14. Spaander MCW, Hoekstra J, Hansen BE, Van Buuren HR, Leebeek FWG, Janssen HLA. Anticoagulant therapy in patients with non-cirrhotic portal vein thrombosis : effect on new thrombotic events and gastrointestinal bleeding. J Thromb Haemost 2013 ; 11 : 452-459.
15. Orr DW, Harrison PM, Devlin J, Karani JB, Kane PA, Heaton ND, et al. Chronic mesenteric venous thrombosis : evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007 ; 5 : 80-86.
16. Randi ML, Tezza F, Scapin M, Duner E, Scarparo P, Scandellari R, et al. Heparin-induced thrombocytopenia in patients with Philadelphia-negative myeloproliferative disorders and unusual splanchnic or cerebral vein thrombosis. Acta Haematol 2010 ; 123 : 140-145.
17. De Gottardi A, Trebicka J, Klinger C, Plessier A, Seijo S, Terziroli B, et al. Antithrombotic treatment with direct-acting oral anticoagulants (DOACs) in patients with splanchnic vein thrombosis and cirrhosis. Liver Int 2016.
18. Sarin SK, Gupta N, Jha SK, Agrawal A, Mishra SR, Sharma BC, et al. Equal efficacy of endoscopic variceal ligation and propranolol in preventing variceal bleeding in patients with noncirrhotic portal hypertension. Gastroenterology 2010 ; 139 : 1238-1245.
19. Guillaume M, Christol C, Plessier A, Corbic M, Péron JM, Sommet A et al. Bleeding risk of variceal band ligation in extrahepatic portal vein obstruction is not increased by oral anticoagulation. Eur J Gastroenterol Hepatol 2018 ; 30 (5) : 563-568.
20. Fanelli F, Angeloni S, Salvatori FM, Marzano C, Boatta E, Merli M, et al. Transjugular intrahepatic portosystemic shunt with expanded-polytetrafuoroethylene-covered stents in non-cirrhotic patients with portal cavernoma. Dig Liver Dis 2011 ; 43 : 78-84.
21. Marot A, Barbosa JV, Denys A, Deltenre P. A new classification of chronic portal vein occlusion for assessing the feasibility of recanalization in non-cirrhotic patients. J Hepatol 2017 ; 66 (suppl. 1) : S 133
22. Duché M, Ducot B, Ackermann O, Guérin F, Jacquemin E, Bernard O. Portal hypertension in children : High-risk varices, primary prophylaxis and consequences of bleeding. J Hepatol 2017 ; 66 : 320-327.
23. Perlemuter G, Béjanin H, Fritsch J, Prat F, Gaudric M, Chaussade S, et al. Biliary obstruction caused by portal cavernoma : a study of 8 cases. J Hepatol 1996 ; 25 (1) : 58-63.
24. Saraswat VA, Rai P, Kumar T, Mohindra S, Dhiman RK. Endoscopic management of portal cavernoma cholangiopathy : practice, principles and strategy. J Clin Exp Hepatol 2014 ; 4 : S67-76.
25. Franceschet I, Zanetto A, Ferrarese A, Burra P, Senzolo M. Therapeutic approaches for portal biliopathy : A systematic review. World J Gastroenterol 2016 ; 22 : 9909.
26. Gauthier-Villars M, Franchi S, Gauthier F, Fabre M, Pariente D, Bernard O. Cholestasis in children with portal vein obstruction. J Pediatr 2005 ; 146 (4) : 568-73.
27. Shneider BL, de Ville de Goyet J, Leung DH, Srivastava A, Ling SC, Duché M, et al. Primary prophylaxis of variceal bleeding in children and the role of MesoRex Bypass : Summary of the Baveno VI Pediatric Satellite Symposium. Hepatol Baltim Md 2016 ; 63 : 1368-1380.
28. Bertocchini A, Falappa P, Grimaldi C, Bolla G, Monti L, de Ville de Goyet J. Intrahepatic portal venous systems in children with noncirrhotic prehepatic portal hypertension : anatomy and clinical relevance. J. Pediatr. Surg. 2014 ; 49 : 1268-1275.
29. De Ville de Goyet J, D’Ambrosio G, Grimaldi C. Surgical Management of Portal Hypertension. Seminars in Pediatric Surgery 2012 ; 21 (3) : 219-232.
30. Abd El-Hamid N, Taylor RM, Marinello D, et al. Aetiology and management of extrahepatic portal vein obstruction in children : King's College Hospital experience. J Pediatr Gastroenterol Nutr 2008 ; 47 (5) : 630-4.
31. Guérin F, Porras J, Fabre M, et al. Liver nodules after portal systemic shunt surgery for extrahepatic portal vein obstruction in children. J Pediatr Surg 2009 ; 44 : 1337-43.
Thrombose de la veine porte au cours de la cirrhose
A. Abergel1, A. Lebreton2, C. Hordonneau3, C. Duron1, F. Nery4
1 Service d’hépato-gastroentérologie, CHU Clermont-Ferrand
2 Service d’hématologie biologique, CHU Clermont-Ferrand
3 Service de radiologie, CHU Clermont-Ferrand
4 Unité de transplantation hépatique et pancréatique, Centre Hospitalier Universitaire de Porto, Portugal
Relecteurs :
C. Francoz5, D. Valla5, S. Hillaire6, E. Rautou7, D. Dutheil8, C. Bureau9, A. Plessier5
5 Service d’hépatologie, APHP Hôpital Beaujon et centre de référence des maladies vasculaires du foie
6 Service de Gastro-entérologie hépatologie, Hôpital Foch, Paris
7 Médecin généraliste, Bagneux
8 Association des malades des vaisseaux du foie (AMVF)
9 Service d’hépato-gastroentérologie, CHU Toulouse
Introduction
La thrombose de la veine porte (TVP) correspond à la présence d’un thrombus, dans la lumière de la veine porte et/ou dans celle des branches portales droite et/ou gauche. L’obstruction du flux sanguin portal qui en résulte peut être partielle ou complète. Le thrombus peut s’étendre aux autres vaisseaux splanchniques, les veines mésentériques inférieure et supérieure et la veine splénique [1]. On distingue classiquement les TVP associées à une cirrhose, les TVP associées à une « thrombophilie » et les envahissements tumoraux de la veine porte, souvent appelés à tort « TVP tumorales » [1].
Facteurs de risque de survenue d’une TVP chez les patients cirrhotiques
Selon la théorie de Virchow, la survenue d’une thrombose vasculaire implique la présence d’un ou plusieurs élément(s) de la triade suivante : hypercoagulabilité, stase veineuse, lésions endothéliales. Ces trois éléments peuvent être impactés chez le patient cirrhotique.
État d’hypercoagulabilité
L’allongement du temps de Quick (TQ) (ou la baisse du taux de prothrombine, TP)) a longtemps conduit à considérer le patient cirrhotique comme à risque hémorragique et « naturellement anticoagulé » [2]. Ces dernières années, des études cliniques et biologiques ont remis en cause ce dogme. Les travaux récents ont démontré que dans certaines conditions expérimentales, il existait même un état d’hypercoagulabilité plasmatique chez le patient cirrhotique [2, 3]. Les principaux mécanismes incriminés sont une diminution acquise de la protéine C, un inhibiteur naturel de la coagulation, associé à une élévation du taux de FVIII. Le lien entre cet état d’hypercoagulabilité et le risque de survenue d’une thrombose a été peu étudié. Une étude cas-témoin a montré que l’augmentation du risque de thrombose veineuse profonde et d’embolie pulmonaire était modeste de l’ordre de 1,7 (1,54-1,95) par rapport à un groupe contrôle sans cirrhose [5]. Une étude rétrospective a par contre montré que la résistance à la thrombomoduline augmentait de façon indépendante (HR = 8,4) le risque de survenue d’une TVP [6]. Une étude prospective est évidemment nécessaire pour conforter ces résultats.
La prévalence des facteurs biologiques de risque de thrombose (innés ou acquis) chez les patients cirrhotiques avec TVP est très variable d’une étude à l’autre [7, 8]. Elle ne semble toutefois pas plus importante que celle observée dans la population générale. La recherche systématique d’anomalies prothrombotiques ne peut pas être recommandée chez les patients cirrhotiques ayant une TVP.
Stase veineuse
Il semblerait que le principal mécanisme favorisant la constitution d’une TVP soit les modifications du flux portal : ralentissement du flux, existence d’un flux en va-et-vient ou son inversion. Les variations du flux portal sont la conséquence des modifications architecturales et de tonus vasculaire associées à la cirrhose [9-14]. Toutefois, le flux est difficile à mesurer en routine, la reproductibilité de la mesure entre les différents appareils ainsi qu’entre opérateurs est faible et doit être améliorée avant de devenir la référence. Enfin, il a été montré que les bêta-bloquants utilisés au cours de l’hypertension portale pourraient provoquer un ralentissement du flux portal et augmenter le risque de TVP [15].
Lésions endothéliales
Il a été suggéré que des lésions endothéliales pré-existantes pourraient favoriser la survenue d’une TVP en favorisant localement l’activation de la coagulation. Les traitements des varices œsophagiennes par sclérothérapie [16], par cyanoacrylate [17] ou du carcinome hépato-cellulaire par injection percutanée d’éthanol [18] ou par radiofréquence [19] pourraient favoriser la survenue d’une TVP.
Épidémiologie, histoire naturelle et classification
La prévalence de la TVP au cours de la cirrhose est très variable dans la littérature. Les séries prospectives les plus récentes suggèrent que la TVP augmente avec la durée d’évolution de la cirrhose et que son incidence à 5 ans est de l’ordre de 10 à 20 % lorsque la TVP est recherchée de façon systématique par une échographie-doppler [2-4]. Les facteurs de risque associent une maladie sévère (cirrhose Child B ou C), un âge avancé et des antécédents d’hémorragie digestive par rupture de varices œso-gastriques [20].
La TVP chez les patients atteints de cirrhose est un événement le plus souvent asymptomatique. Dans plus de deux tiers des cas, la veine porte se reperméabilise spontanément, d’autant plus fréquemment que la thrombose est partielle [21]. À l’inverse, l’extension de la thrombose aux autres vaisseaux splanchniques survient, selon les séries, dans 6 à 48 % des cas. Alors que la TVP a longtemps été considérée comme un élément péjoratif au cours de l’histoire naturelle de la cirrhose, les études les plus récentes suggèrent que la TVP n’augmente pas le risque de décompensation et n’a pas d’influence sur la survie [2, 5, 6].
La classification de la TVP la plus utilisée est la classification anatomique de Yerdel. [22] Plus récemment, Sarin et al. ont proposé une nouvelle classification [1], qui prend en compte non seulement la localisation et l’extension du thrombus, mais également l’ancienneté de la thrombose, son mode de présentation et la maladie hépatique sous-jacente. L’objectif de cette classification (tableau 1) est d’harmoniser la prise en charge et de pouvoir comparer les études à venir entre elles.
| Localisation de la TVP – (Type 1, 2a, 2b, 3). |
| – Type 1 : Au niveau du tronc uniquement– Type 2 : Au niveau de(s) branche(s) uniquement : 2a : une branche 2b : les deux branches– Type 3 : Au niveau du tronc et des branches |
| Degré d’occlusion du système veineux portal (O, NO) |
| – O : Occlusive : aucun flux visible dans la lumière de la veine porte à l’imagerie Doppler– NO : Non occlusive : flux visible dans la lumière de la veine porte par imagerie Doppler |
| Ancienneté et presentation (R, C) |
| – R : Récente (détectée pour la première fois sur une veine porte au préalable perméable (non thrombosé), présence d’un thrombus hyperdense à l’imagerie, circulation collatérale absente ou limitée, VP dilatée au niveau de l’occlusion) : - Asymptomatique (As) - Symptomatique (S) : Caractéristiques de la TVP aiguës (avec ou sans ischémie intestinale aiguë)– Ch : Chronique (pas de thrombus hyperdense, préalablement diagnostiquée lors du suivi d’une TVP, cavernome portal et manifestations cliniques de l’hypertension portale (HTP)) : - Asymptomatique - Symptomatique : caractéristiques de l’hypertension portale (avec ou sans HTP) |
| Extension du thrombus (S, M, SM) |
| – Veine splénique (S),– Veine mésentérique (M)– ou les deux (SM) |
| Type et présence de maladie hépatique sous-jacente |
| – Cirrhotique– Maladie hépatique non cirrhotique,– Après une transplantation hépatique– Carcinome hépatocellulaire (CHC)– Tumeurs malignes locales– Et affections associées |
Tableau 1
Classification anatomo-fonctionnelle de la TVP au cours de la cirrhose, de Sarin et al. [1].
Diagnostic
La TVP au cours de la cirrhose est le plus souvent découverte de façon fortuite. Les signes cliniques qui peuvent conduire au diagnostic sont non spécifiques et incluent une fièvre, des douleurs abdominales, des nausées et/ou des vomissements, une diarrhée parfois sanglante pouvant alors correspondre à une ischémie veineuse mésentérique associée. La TVP peut également être révélée à l’occasion d’une rupture de varices gastriques ou œsophagiennes [8]. Il n’existe aucune corrélation entre la présence de symptômes et l’extension de la thrombose [9]. Aucun examen biologique n’oriente vers le diagnostic de TVP. L’examen de première intention réalisé devant toute suspicion de TVP est l’échographie-doppler. Sa sensibilité est très bonne en cas de TVP complète mais beaucoup plus faible (50 %) lorsque la thrombose est partielle ou lorsqu’il s’agit d’une thrombose n’atteignant que la veine splénique ou la veine mésentérique supérieure [23, 24]. L’échographie-doppler doit obligatoirement être complétée par une tomodensitométrie (TDM) ou une IRM avec injection de produit de contraste (excellentes sensibilité et spécificité pour le diagnostic de TVP) [25]. Ces examens permettent (1) de préciser les arguments en faveur d’une cirrhose ainsi que de ses complications (foie à contours bosselés, dysmorphie hépatique avec atrophie du segment IV, ascite, voies de dérivations porto-systémiques), (2) de confirmer la nature fibrinocruorique du thrombus et (3) de préciser l’extension du thrombus dans les différents vaisseaux splanchniques ainsi que les conséquences ischémiques éventuelles que la thrombose peut avoir sur le tube digestif. Le diagnostic différentiel entre thrombus fibrinocruorique et obstruction portale tumorale est parfois difficile et nécessite une relecture spécialisée. Les arguments pour une obstruction tumorale sont un élargissement vasculaire et une prise de contraste endovasculaire à la phase artérielle. L’existence d’un nodule évocateur de carcinome hépatocellulaire à proximité de la thrombose ainsi que l’élévation de l’alpha-fœtoprotéine sérique sont en faveur d’une obstruction tumorale. Dans les cas difficiles, l’échographie de contraste peut aider (prise de contraste intraluminale évocatrice d’obstruction tumorale) et, en dernier recours, une biopsie peut être réalisée. De même, lorsque le diagnostic de cirrhose n’est pas certain, il est important de colliger les arguments pour ou contre une cirrhose en présence d’une thrombose porte. Dans une étude espagnole, la mesure de l’élasticité hépatique était significativement plus basse au cours de la thrombose porte sans cirrhose, (6,4 ± 2,2 kPa), par rapport à un groupe contrôle de patients avec cirrhose (40,9 ± 20,5 kPa) [26]. Les signes orientant vers une souffrance digestive [23-25] sur le scanner sont, en association à une occlusion distale de la veine mésentérique supérieure, des anomalies pariétales intestinales incluant un épaississement pariétal étendu avec œdème sous-muqueux, une infiltration du mésentère et un épanchement intra-abdominal [27].
Traitement
Les antagonistes de la vitamine K (AVK) ou une héparine de bas poids moléculaires peuvent être utilisés. Il faut toutefois souligner que les indicateurs habituels d’efficacité thérapeutique (INR (International Normalized Ratio) ou dosage de l’activité anti-Xa) chez les patients sans maladie du foie sont difficilement interprétables chez les patients avec une insuffisance hépatique.
L’efficacité des anticoagulants d’une part, et le risque hémorragique d’autre part, au cours de la TVP du cirrhotique ont été évalués dans de nombreuses séries. Les résultats d’une méta-analyse récente ont montré que le taux de recanalisation était de l’ordre de 70 % sous anticoagulants, comparé à 40 % sans traitement et que les anticoagulants diminuaient le risque d’extension de la thrombose (10 % chez les patients traités, contre 33 % chez les malades non traités). De plus, les anticoagulants n’exposaient pas les patients à un sur-risque hémorragique [28].
Le traitement anticoagulant est clairement indiqué chez les patients cirrhotiques avec TVP et candidats à une transplantation hépatique, l’objectif étant de recanaliser tout ou partie de la veine porte. En effet, l’existence d’une thrombose porte complique la transplantation hépatique en rendant difficile l’anastomose de la veine porte du greffon sur le territoire splanchnique du receveur. Les techniques alternatives sont plus complexes, et augmentent la morbidité et la mortalité après transplantation. En cas d’obstruction complète du système porte, la transplantation hépatique peut parfois être contre-indiquée [30].
Chez les patients non candidats à une transplantation hépatique, il n’y a pas d’indication d’anticoagulation en dehors de l’ischémie mésentérique [29]. Néanmoins, il a été également suggéré que l’administration prolongée d’anticoagulants pourrait diminuer le risque de décompensation et améliorer la survie chez des patients ayant une cirrhose sans TVP [31].
Dans deux études récentes chez des patients cirrhotiques, les anticoagulants oraux directs (rivaboxaban et apixaban) étaient aussi efficaces pour prévenir la récidive d’un accident vasculaire cérébral que les AVK et les héparines de bas poids moléculaire (HBPM), sans majorer le risque hémorragique, ni celui de défaillance hépatique [32, 33]. Comme en population générale, le taux d’hémorragies graves était moins important sous anticoagulants oraux directs (AOD) que sous anticoagulation standard. Chez les patients porteurs d’une cirrhose peu sévère (Child-Pugh A), il y a suffisamment de littérature pour recommander les AOD dans l’AC/FA (arythmie cardiaque par fibrillation auriculaire) et les thromboses veineuses profondes (phlébite, embolie pulmonaire). Le niveau de preuve est cependant insuffisant pour formuler des recommandations concernant les AOD chez les patients avec une insuffisance hépatique (Child-Pugh B et C).
Le TIPS (Transjugular Intrahepatic Portosystemic Shunt) a longtemps été contre-indiqué chez les patients cirrhotiques avec TVP. Toutefois, il a été montré récemment que le TIPS, pouvait aider à recanaliser tout ou partie de la veine porte et rétablir un flux portal satisfaisant. [34]. Une étude contrôlée randomisée a montré que dans une population très sélectionnée de patients cirrhotiques avec TVP ayant fait une hémorragie digestive par rupture de varices, le TIPS augmentait les chances de recanalisation de la veine porte sans augmenter le risque d’encéphalopathie ni de décès, comparé au traitement de référence : endoscopique + bêta-bloquants [35]. Ces résultats doivent toutefois être confirmés dans de plus grandes populations. À l’heure actuelle, il n’y a pas d’indication de TIPS pour une TVP isolée. Le TIPS peut être proposé chez les patients ayant une ascite réfractaire ou une hémorragie digestive non contrôlée par les traitements habituels [29], et en cas d’echec de reperméabilisation sous traitement anticoagulant ou en cas d’accident hémorragique sous anticoagulant.
Le TIPS est un traitement à réserver et à discuter en deuxième ligne après avis auprès d’un centre de compétence/référence dans les maladies vasulaires du foie et avec l’expertise du TIPS dans cette situation.
Conclusion
De nombreux progrès ont été faits au cours des dernières années concernant les connaissances sur la TVP au cours de la cirrhose. À l’heure actuelle, peu d’études prospectives ou de travaux multicentriques ont été réalisés. Ceux-ci pourraient permettre, dans un avenir proche, d’améliorer nos connaissances sur le sujet, en particulier de mieux identifier les patients à risque de développer une TVP, d’affiner les indications de traitement anticoagulant ainsi que de déterminer la place des anticoagulants oraux directs.
Recommandations
- •Faire une échographie-doppler en première intention pour dépister une thrombose de la veine porte chez un patient atteint de cirrhose (C1).
- •Faire un scanner 4 temps, pour confirmer le diagnostic, évaluer l’extension, et éliminer un envahissement tumoral (A1).
- •À chaque échographie semestrielle de dépistage du CHC, une thrombose de la veine porte doit être recherchée par un examen doppler (A1).
- •Traiter avec une prophylaxie adaptée l’HTP, en cas de thrombose porte (B2).
- •L’intérêt du bilan de « thrombophilie » chez le patient cirrhotique porteur d’une TVP n’est pas établi (C2).
- •Traiter tout patient avec une ischémie mésentérique (A1) par anticoagulant pendant au moins 6 mois.
- •Traiter les patients cirrhotiques avec TVP en liste d’attente pour la greffe hépatique par HBPM ou AVK jusqu’à la greffe hépatique (B2).
- •Discuter un traitement anticoagulant chez les patients cirrhotiques avec TVP et sans contre-indication définitive à la greffe (C2).
- •Le niveau de preuve est trop faible pour recommander un anticoagulant par rapport à un autre (C2).
- •Envisager un TIPS en cas de complication de l’HTP chez un patient ayant une cirrhose porteur d’une thrombose porte (B1).
- •Informer les patients de l’existence des associations de patients dès l’annonce du diagnostic.
Références
1. Sarin SK, Philips CA, Kamath PS, Choudhury A, Maruyama H, Nery FG, et al. Toward a Comprehensive New Classification of Portal Vein Thrombosis in Patients With Cirrhosis. Gastroenterology 2016 ; 151 (4) : 574-7 e3.
2. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med 2011 ; 365 (2) : 147-56.
3. Lebreton A, Sinegre T, Pereira B, Lamblin G, Duron C, Abergel A. Plasma hypercoagulability in the presence of thrombomodulin but not of activated protein C in patients with cirrhosis. J Gastroenterol Hepatol 2017 ; 32 (4) : 916-24.
4. Tripodi A, Primignani M, Lemma L, Chantarangkul V, Mannucci PM. Evidence that low protein C contributes to the procoagulant imbalance in cirrhosis. J Hepatol 2013 ; 59 (2) : 265-70.
5. Søgaard KK, Horváth-Puhó E, Grønbaek H, Jepsen P, Vilstrup H, Sørensen HT. Risk of venous thromboembolism in patients with liver disease : a nationwide population-based case-control study. Am J Gastroenterol 2009 ; 104 (1) : 96-101.
6. La Mura V, Tripodi A, Tosetti G, Cavallaro F, Chantarangkul V, Colombo M, et al. Resistance to thrombomodulin is associated with de novo portal vein thrombosis and low survival in patients with cirrhosis. Liver Int 2016 ; 36 (9) : 1322-30.
7. Qi X, De Stefano V, Su C, Bai M, Guo X, Fan D. Associations of antiphospholipid antibodies with splanchnic vein thrombosis : a systematic review with meta-analysis. Medicine (Baltimore) 2015 ; 94 (4) : e496.
8. Qi X, Ren W, De Stefano V, Fan D. Associations of coagulation factor V Leiden and prothrombin G20210A mutations with Budd-Chiari syndrome and portal vein thrombosis : a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2014 ; 12 (11) : 1801-12 e7.
9. Ma J, Yan Z, Luo J, Liu Q, Wang J, Qiu S. Rational classification of portal vein thrombosis and its clinical significance. PLoS One 2014 ; 9 (11) : e112501.
10. Zocco MA, Di Stasio E, De Cristofaro R, Novi M, Ainora ME, Ponziani F, et al. Thrombotic risk factors in patients with liver cirrhosis : correlation with MELD scoring system and portal vein thrombosis development. J Hepatol 2009 ; 51 (4) : 682-9.
11. Abdel-Razik A, Mousa N, Elhelaly R, Tawfik A. De-novo portal vein thrombosis in liver cirrhosis : risk factors and correlation with the Model for End-stage Liver Disease scoring system. Eur J Gastroenterol Hepatol 2015 ; 27 (5) : 585-92.
12. Chen H, Turon F, Hernández-Gea V, Fuster J, Garcia-Criado A, Barrufet M, et al. Nontumoral portal vein thrombosis in patients awaiting liver transplantation. Liver Transpl 2016 ; 22 (3) : 352-65
13. Nery F, Chevret S, Condat B, de Raucourt E, Boudaoud L, Rautou PE, et al. Causes and consequences of portal vein thrombosis in 1,243 patients with cirrhosis : results of a longitudinal study. Hepatology 2015 ; 61 (2) : 660-7.
14. Stine JG, Wang J, Shah PM, Argo CK, Intagliata N, Uflacker A, Caldwell SH, Northup PG. Decreased portal vein velocity is predictive of the development of portal vein thrombosis : A matched case-control study. Liver Int 2018 ; 38 (1) : 94-101.
15. Qi XS, Bai M, Fan DM. Nonselective beta-blockers may induce development of portal vein thrombosis in cirrhosis. World J Gastroenterol. 2014 ; 20 (32) : 11463-6.
16. Amitrano L, Brancaccio V, Guardascione MA, Margaglione M, Sacco M, Martino R, et al. Portal vein thrombosis after variceal endoscopic sclerotherapy in cirrhotic patients : role of genetic thrombophilia. Endoscopy 2002 ; 34 (7) : 535-8.
17. Chang CJ, Shiau YT, Chen TL, Hou MC, Sun CH, Liao WC, Lin HC, Lee SD. Pyogenic portal vein thrombosis as a reservoir of persistent septicemia after cyanoacrylate injection for bleeding gastric varices. Digestion. 2008 ; 78 (2-3) : 139-43.
18. Habu D, Nishiguchi S, Shiomi S, Tamori A, Sakaguchi H, Takeda T, et al. Portal vein thrombosis following percutaneous ethanol injection therapy for hepatocellular carcinoma. Indian J Gastroenterol 2002 ; 21 : 162-3.
19. Francica G. Complications of radio-frequency thermal ablation. Radiology 2001 ; 220 : 554.
20. Maruyama H, Okugawa H, Takahashi M, Yokosuka O. De novo portal vein thrombosis in virus-related cirrhosis : predictive factors and long-term outcomes. Am J Gastroenterol 2013 ; 108 (4) : 568-
21. Luca A, Caruso S, Milazzo M, Marrone G, Mamone G, Crino F, et al. Natural course of extrahepatic nonmalignant partial portal vein thrombosis in patients with cirrhosis. Radiology 2012 ; 265 : 124-32.
22. Yerdel MA, Gunson B, Mirza D, Karayalcin K, Olliff S, Buckels J, et al. Portal vein thrombosis in adults undergoing liver transplantation : risk factors, screening, management, and outcome. Transplantation 2000 ; 69 (9) : 1873-81.
23. Singal AK, Kamath PS, Tefferi A. Mesenteric venous thrombosis. Mayo Clin Proc 2013 ; 88 : 285-94.
24. Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001 ; 345 (23) : 1683-8.
25. Margini C, Berzigotti A. Portal vein thrombosis : The role of imaging in the clinical setting. Dig Liver Dis 2017 ; 49 (2) : 113-20.
26. Seijo S, Reverter E, Miquel R, Berzigotti A, Abraldes JG, Bosch J, García-Pagán JC. Role of hepatic vein catheterisation and transient elastography in the diagnosis of idiopathic portal hypertension. Dig Liver Dis 2012 ; 44 (10) : 855-60.
27. Elkrief L, Corcos O, Bruno O, Larroque B, Rautou PE, Zekrini K, et al. Type 2 diabetes mellitus as a risk factor for intestinal resection in patients with superior mesenteric vein thrombosis. Liver Int 2014 ; 34 (9) : 1314-21.
28. Loffredo L, Pastori D, Farcomeni A, Violi F. Effects of Anticoagulants in Patients With Cirrhosis and Portal Vein Thrombosis : a Systematic Review and Meta-Analysis. Gastroenterology 2017 ; 153 : 480-7.
29. European Association for the Study of the Liver. EASL Clinical Practice Guidelines : Vascular diseases of the liver. J Hepatol 2016 ; 64 (1) : 179-202.32.
30. Francoz C, Valla D, Durand F. Portal vein thrombosis, cirrhosis, and liver transplantation. J Hepatol 2012 ; 57 (1) : 203-12.
31. Villa E, Cammà C, Marietta M, Luongo M, Critelli R, Colopi S, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012 ; 143 (5) : 1253-60.
32. Hum J, Shatzel JJ, Jou JH, Deloughery TG. The efficacy and safety of direct oral anticoagulants vs traditional anticoagulants in cirrhosis. Eur J Haematol 2017 ; 98 : 393-7.
33. Intagliata NM, Henry ZH, Maitland H, Shah NL, Argo CK, Northup PG, et al. Direct Oral Anticoagulants in Cirrhosis Patients Pose Similar Risks of Bleeding When Compared to Traditional Anticoagulation. Dig Dis Sci 2016 ; 61 (6) : 1721-7.
34. Zhao M, Yue Z, Zhao H, Wang L, Fan Z, He F, et al. Techniques of TIPS in the treatment of liver cirrhosis combined with incompletely occlusive main portal vein thrombosis. Sci Rep 2016 ; 6 : 33069.
35. Luo X, Wang Z, Tsauo J, Zhou B, Zhang H, Li X. Advanced Cirrhosis Combined with Portal Vein Thrombosis : A Randomized Trial of TIPS versus Endoscopic Band Ligation Plus Propranolol for the Prevention of Recurrent Esophageal Variceal Bleeding. Radiology 2015 ; 276 (1) : 286-93.
Maladie vasculaire porto-sinusoïdale
P. Bedossa1 et P.E. Rautou2
1 Anatomo-Pathologie, APHP Hôpital Beaujon
2 Service d’hépatologie, APHP Hôpital Beaujon
Relecteurs :
O. Goria3, D. Valla4, V. Lambert5, D. Dutheil6, C. Bureau7, A. Plessier4
3 Service d’hépato-gastroentérologie, CHU Rouen
4 Service d’hépatologie, APHP Hôpital Beaujon et centre de référence des maladies vasculaires du foie
5 Médecin généraliste, Angers
6 Association des malades des vaisseaux du foie (AMVF)
7 Service d’hépato-gastroentérologie, CHU Toulouse
Introduction
Le terme maladie vasculaire porto-sinusoïdale (MVPS) regroupe diverses maladies caractérisées par des anomalies des petits vaisseaux du foie précédemment nommées d’un point de vue histologique « hyperplasie nodulaire régénérative », « veinopathie portale oblitérante », « sclérose hépatoportale », « fibrose septale incomplète », « fibrose portale non cirrhotique » ou d’un point de vue clinique « hypertension portale idiopathique », « hypertension portale intrahépatique non cirrhotique ». En effet, il y a un fort chevauchement entre toutes ces entités et à ce jour pas de conséquence pratique à les discriminer (tableau 1) [1].
| Entité | Définition |
| Hypertension portale intrahépatique non cirrhotique | Augmentation de la pression porte due à des lésions hépatiques autres qu’une cirrhose, avec des veines porte et hépatiques perméables |
| Hypertension portale non cirrhotique | Augmentation de la pression porte due à des lésions hépatiques autres qu’une cirrhose due à des lésions intra ou pré-hépatiques |
| Hypertension portale idiopathique | Maladie de cause inconnue caractérisée par une splénomégalie, une anémie et une hypertension portale, en l’absence de cirrhose, de maladie hématologique, de parasite, d’occlusion des veines hépatiques ou de la veine porte, de granulome, ou de fibrose hépatique congénitale |
| Fibrose portale non cirrhotique | Hypertension portale idiopathique associée à des quantités variables de fibrose intrahépatique localisée essentiellement autour des espaces portes |
| Hyperplasie nodulaire régénérative | Transformation diffuse micronodulaire du parenchyme hépatique sans fibrose entre les nodules |
| Veinopathie portale oblitérante | Épaississement de l’intima avec rétrécissement de la lumière des branches portes intrahépatiques de gros, moyen ou petit calibre, en l’absence de cirrhose et de thrombose porte extrahépatique |
| Sclérose hépatoportale | Épaississement fibreux de la veine porte et de ses branches chez des malades avec hypertension portale non cirrhotique |
| Cirrhose septale incomplète | Cirrhose macronodulaire avec des septa fins et souvent incomplets qui délimitent des nodules de grande taille difficilement visibles |
Tableau 1
Définitions des différentes entités maintenant regroupées sous le terme de maladie vasculaire porto-sinusoïdale [1].
Il est en revanche utile de bien différencier la MVPS du syndrome d’obstruction sinusoïdal (SOS) et de maladie veino-occlusive (MVO). Ces lésions sont primitivement sinusoïdales s’étendant vers la veine centrolobulaire. Elles ont des causes différentes (toxicité des dérivés des alcaloïdes de la pyrozilidine, conditionnement de greffe de moelle, oxaplatine) et une présentation clinique distincte avec notamment une plus grande fréquence de l’insuffisance hépatique. Le syndrome d’obstruction sinusoïdal et la maladie veino-occlusive ne seront pas abordés ici.
Le diagnostic de MVPS est défini selon le Réseau européen des maladies vasculaires du foie VALDIG [2], par une des trois associations suivantes (tableau 2) :
- a)absence de cirrhose prouvée sur une biopsie hépatique adéquate et au moins un signe spécifique d’hypertension portale ;
- b)absence de cirrhose prouvée sur une biopsie hépatique adéquate et au moins un signe histologique spécifique de MVPS ;
- c)absence de cirrhose prouvée sur une biopsie hépatique adéquate, et au moins un signe non spécifique d’hypertension portale et au moins un signe histologique non spécifique de MVPS.
| Signes d’hypertension portale | Signes histologiques spécifiques de MVPS | |
| Spécifiques | – Varices gastriques, œsophagiennes ou ectopiques– Hémorragie digestive par hypertension portale– Collatérales porto-systémiques en imagerie | – Veinopathie portale oblitérante (épaississement de la paroi veineuse portale, occlusion de la lumière veineuse portale, disparition des veinules portes)– Hyperplasie nodulaire régénérative– Fibrose/cirrhose septale incomplète |
| Non spécifiques | – Ascite– Taux de plaquettes < 150 000/mm3– Taille de rate ≥ 13 cm dans le plus grand axe | – Anomalies des espaces portes (multiplication, dilatation des artérioles, vaisseaux périportaux, vaisseaux aberrants)– Architectural désorganisée : distribution irrégulière des espaces portes et des veines centrolobulaires– Dilatation sinusoïdale non zonale– Fibrose périsinusoïdale légère |
Tableau 2
Critères à prendre en compte pour le diagnostic de MVPS.
Diagnostic
Quand évoquer le diagnostic de MVPS ?
Les deux modes de découvertes principaux de la MVPS sont des anomalies inexpliquées des tests hépatiques et une hypertension portale sans cirrhose. Dans les deux cas, l’attention doit être attirée par l’absence de cause reconnue d’hépatopathie chronique, telles qu’une consommation excessive de boissons alcoolisées, un syndrome métabolique, une infection par le virus de l’hépatite B ou de l’hépatite C, une hépatite auto-immune ou une obstruction des veines hépatiques.
Les altérations des tests hépatiques associées à la MVPS sont variées. Il s’agit chez les deux tiers des malades d’une élévation des transaminases le plus souvent modéréé, chez la moitié des malades d’une élévation des phosphatases alcalines, parfois supérieures à 2 fois la limite supérieure de la normale, et chez les trois quarts des malades d’une élévation des γ-GT [3, 4]. Dans deux études récentes ayant inclus plus de 1 200 malades avec anomalies inexpliquées du bilan hépatique, des lésions de MVPS étaient présentes dans 15 à 20 % des cas [5, 6]. Ces lésions de MVPS étaient fréquemment isolées, sans hypertension portale.
L’autre manifestation de la MVPS est l’hypertension portale (tableau 2).
Outre l’absence de cause d’hépatopathie, les autres signes qui doivent alerter et pousser les investigations sont un contraste entre une hypertension portale franche et des tests de fonction hépatique normaux ou quasi normaux. Ainsi, le taux de prothrombine est généralement supérieur à 50 % chez les malades avec MVPS. L’autre tableau qui doit alerter est le contraste entre les signes d’hypertension portale et une élasticité hépatique basse (typiquement FibroScan® hépatique < 10 kPa mais aucun seuil validé à ce jour) [7].
Il faut noter que la thrombose de la veine porte est une conséquence de la MVPS possiblement en raison du ralentissement du flux porte induit par le bloc intrahépatique. Cependant, une obstruction porte chronique peut aussi causer des remaniements du parenchyme hépatique proche d’une MVPS. Ainsi, en pratique, devant un malade ayant une thrombose porte ancienne, il n’est pas possible de déterminer si la MVPS est la cause ou la conséquence de la thrombose porte.
Comment confirmer le diagnostic de MVPS ?
Une confrontation des données cliniques avec les résultats anatomopathologiques et radiologiques est nécessaire. La biopsie hépatique peut être réalisé par voie transpariétale ou transjugulaire pour confirmer le diagnostic de maladie porto-sinusoïdale.
Une biopsie hépatique est considérée comme adéquate si elle mesure plus de 20 mm de long et contient plus de 10 espaces portes et est peu/pas fragmentée ou bien est considérée adéquate par un anatomopathologiste expert.
Si la biopsie est effectuée par voie transjugulaire, le gradient de pressions veineuses hépatiques peut être mesuré. En raison du bloc présinusoïdal, en cas de MVPS, le gradient de pressions veineuses hépatiques mesuré par voie transjugulaire n’est pas corrélé au gradient porto-systémique [8], ce qui a deux conséquences : (a) un gradient de pressions veineuses hépatiques à moins de 10 mmHg n’élimine pas la présence de varices gastro-œsophagiennes ; (b) un gradient de pressions hépatiques à moins de 10 mmHg chez un malade ayant des signes évidents d’hypertension portale constitue un argument de plus en faveur d’une MVPS. Des voies de dérivations veino-veineuses hépatiques doivent être systématiquement recherchées puisqu’elles sont souvent présentes et qu’elles faussent l’interprétation de la mesure de pression de la veine hépatique bloquée.
Les signes histologiques spécifiques et non spécifiques sont rapportés dans le tableau 2.
Associations étiologiques
De nombreuses affections ont été rapportées associées à l’existence d’une MVPS. Elles sont résumées dans le tableau 3.
| Infections :– VIH– Angiocholites à répétition (anastomose bilio-digestive) |
| Maladies dysimmunitaires ou de système :– Déficit immun commun variable– Maladie de Basedow– Polyarthrite rhumatoïde– Syndrome POEMS– Lupus érythémateux disséminé– Maladie de Wegener– Syndrome de Sharp |
| Maladies hématologiques :– Myélome multiple– Maladie de Waldenstrom– Syndrome myélodysplasique– Syndrome myéloprolifératif– Maladie de Hodgkin– Lymphome B marginal– Purpura idiopathique thrombocytopénique |
| États prothrombotiques :– Mutation du gène du facteur II– Mutation du gène du facteur V– Syndrome des anticorps antiphospholipdes– Déficit en protéine S– Déficit en protéine C |
| Médicaments (tous débatus) :– Didanosine– Azathioprine,– 6-thioguanine,– Arsenic |
| Maladies génétiques :– Syndrome d’Adams-Oliver– Syndrome de Turner– Mutation des gènes des télomérases (TERT/TERC)– Formes familiales– Mucovicidose |
Tableau 3
États associés aux MVPS [3, 4, 13].
Histoire naturelle
L’histoire naturelle des formes de MVPS sans hypertension portale n’est pas connue. Il est possible qu’une proportion importante de ces malades ne développe jamais de signes d’hypertension portale. Ainsi, deux séries autopsiques ont trouvé une prévalence de l’hyperplasie nodulaire régénérative de 2 à 3 % en population générale et la grande majorité de ces malades n’avaient pas de signes d’hypertension portale [6].
L’histoire naturelle des formes avec hypertension portale est mieux connue. L’hémorragie digestive par rupture de varices est l’événement clinique le plus fréquent, touchant près de la moitié des malades qui ne reçoivent pas de prophylaxie. Même lorsqu’une prophylaxie de la rupture de varices œsophagiennes est mise en place, le risque à 5 ans d’hémorragie digestive est de 25 %. La mortalité causée par une hémorragie digestive par rupture de varices demeure faible, soit 3 % à 6 semaines [4, 9].
L’ascite n’est habituellement pas présente et survient le plus souvent au cours d’événements intercurrents tels qu’une infection ou une hémorragie digestive. Sa présence témoigne d’un stade plus avancé de la maladie puisqu’elle était associée à un risque accru de décès [9].
L’encéphalopathie hépatique est une manifestation encore plus rare et elle est habituellement causée par des dérivations porto-systémiques de large calibre.
Le syndrome hépato-pulmonaire pourrait toucher jusqu’à 10 % des malades.
L’évolution vers une insuffisance hépatique avancée n’est pas habituelle, mais est possible.
La thrombose porte est fréquente en cas de MVPS avec hypertension portale, touchant un tiers des malades à 5 ans. Ceux qui ont une infection VIH (virus de l’immunodéficience humaine) ou dont le diagnostic de maladie du foie a été révélé par une rupture de varices sont particulièrement à risque [4]. Étant donné cette incidence, une surveillance radiologique de la perméabilité de la veine porte peut être proposée même s’il n’existe pas de littérature appuyant directement cette pratique.
Contrairement à la cirrhose, la survenue d’un carcinome hépato-cellulaire est exceptionnelle en cas de MVPS.
Au long terme, la survie à 10 ans des malades avec MVPS et hypertension portale va de 56 à 82 %. La mortalité est liée aux maladies extrahépatiques associées à la MVPS plutôt qu’aux complications de la maladie hépatique [9].
Prise en charge thérapeutique
Dans les formes de MVPS sans hypertension portale, l’évolution n’étant pas connue, une simple surveillance peut être proposée. Cette surveillance biologique, par imagerie, voire par endoscopie, a pour but de dépister la survenue de signes d’hypertension portale qui modifieraient alors le suivi et la prise en charge.
Chez les malades avec hypertension portale, en l’absence d’étude concernant spécifiquement cette maladie, on propose que les varices gastro-œsophagiennes soient prises en charge comme chez les malades avec cirrhose [10, 11].
La mise en place d’une prothèse de dérivation porto-cave par voie trans-jugulaire (TIPS) peut être envisagée pour les malades avec hémorragie digestive non contrôlée par les traitements médicaux et endoscopiques. Les résultats sont alors favorables [8]. Lorsqu’il existe une ascite réfractaire, les résultats du TIPS sont moins bons, surtout lorsque la créatininémie est supérieure à 100 μmol/L et/ou qu’il y a des comorbidités importantes [8].
La transplantation hépatique est une option pour les malades avec ascite réfractaire ou insuffisance hépato-cellulaire avancée en l’absence de contre-indication. La survie à long terme rapportée après transplantation hépatique est bonne [12].
Étant donné la présence fréquente d’un état prothrombotique et la survenue d’une thrombose porte dans l’histoire naturelle de la maladie, un traitement systématique par anticoagulation a été évoqué, surtout lorsqu’un état procoagulant est mis en évidence. Cette stratégie mérite d’être évaluée dans des études prospectives.
Recommandations
- •Le terme « maladie vasculaire porto-sinusoïdale » recouvre les entités précédemment nommées hyperplasie nodulaire régénérative, veinopathie portale oblitérante, sclérose hépatoportale, fibrose septale incomplète, fibrose portale non cirrhotique, hypertension portale idiopathique, hypertension portale intrahépatique non cirrhotique (C2).
- •Évoquer une maladie vasculaire porto-sinusoïdale dans les situations suivantes : hypertension portale avec tests de fonction hépatique normaux ou quasi normaux ; hypertension portale avec élasticité hépatique basse ; anomalies inexpliquées des tests hépatiques même sans hypertension portale (B1).
- •Une élasticité hépatique basse ou un gradient de pressions hépatiques < 10 mmHg n’éliminent pas la présence de varices gastro-œsophagiennes en cas de maladie vasculaire porto-sinusoïdale (B2).
- •Une biopsie hépatique de bonne qualité (> 20 mm) montrant l’absence de cirrhose est essentielle au diagnostic (C2).
- •Chercher les affections qui ont été décrites comme associées à la maladie vasculaire porto-sinusoïdale (B1).
- •Dépister les varices gastro-œsophagiennes par endoscopie digestive haute au diagnostic de maladie vasculaire porto-sinusoïdale. Au cours du suivi, la fréquence des endoscopies digestives hautes de dépistage des varices n’est pas codifiée (B1).
- •Dans le suivi de la maladie vasculaire porto-sinusoïdale avec signes d’hypertension portale, dépister la survenue de thrombose porte par une échographie couplée au doppler hépatique tous les six mois (B1).
- •Aucune recommandation ne peut être donnée concernant l’anticoagulation en prophylaxie de la thrombose porte (C2).
- •Informer les patients de l’existence des associations de patients dès l’annonce du diagnostic.
Références
1. Plessier A, Rautou PE, Valla DC. Management of hepatic vascular diseases. J Hepatol 2012 ; 56 (Suppl. 1) : S25-38.
2. Valla DC, Cazals-Hatem D. Vascular liver diseases on the clinical side : definitions and diagnosis, new concepts. Virchows Arch 2018 ; 473 (1) : 3-13.
3. Cazals-Hatem D, Hillaire S, Rudler M, Plessier A, Paradis V, Condat B, Francoz C, et al. Obliterative portal venopathy : Portal hypertension is not always present at diagnosis. J Hepatol 2011 ; 54 (3) : 455-61.
4. Siramolpiwat S, Seijo S, Miquel R, Berzigotti A, Garcia-Criado A, Darnell A, et al. Idiopathic portal hypertension : natural history and long-term outcome. Hepatology 2014 ; 59 : 2276-2285.
5. Barge S, Grando V, Nault JC, Broudin C, Beaugrand M, Ganne-Carrie N, et al. Prevalence and clinical significance of nodular regenerative hyperplasia in liver biopsies. Liver Int 2016 ; 36 : 1059-1066.
6. Elkrief L, Rautou PE. Idiopathic non-cirrhotic portal hypertension : the tip of the obliterative portal venopathies iceberg? Liver Int 2016 ; 36 : 325-327.
7. Seijo S, Reverter E, Miquel R, Berzigotti A, Abraldes JG, Bosch J, Garcia-Pagan JC. Role of hepatic vein catheterisation and transient elastography in the diagnosis of idiopathic portal hypertension. Dig Liver Dis 2012 ; 44 : 855-860.
8. Bissonnette J, Garcia-Pagan JC, Albillos A, Turon F, Ferreira C, Tellez L, et al. Role of the Transjugular Intrahepatic Portosystemic Shunt in the Management of Severe Complications of Portal Hypertension in Idiopathic Noncirrhotic Portal Hypertension. Hepatology 2016 ; 64 (1) : 224-31.
9. Schouten JN, Nevens F, Hansen B, Laleman W, van den Born M, Komuta M, et al. Idiopathic noncirrhotic portal hypertension is associated with poor survival : results of a long-term cohort study. Aliment Pharmacol Ther 2012 ; 35 : 1424-1433.
10. de Franchis R. Expanding consensus in portal hypertension : Report of the Baveno VI Consensus Workshop : Stratifying risk and individualizing care for portal hypertension. J Hepatol 2015 ; 63 : 743-752.
11. European Association for the Study of the Liver. Electronic address eee. EASL Clinical Practice Guidelines : Vascular diseases of the liver. J Hepatol 2016 ; 64 : 179-202.
12. Radomski JS, Chojnacki KA, Moritz MJ, Rubin R, Armenti VT, Wilson GA, et al. Results of liver transplantation for nodular regenerative hyperplasia. Am Surg 2000 ; 66 : 1067-1070.
13. Schouten JN, Garcia-Pagan JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology 2011 ; 54 : 1071-1081.
Syndrome d’obstruction sinusoïdale
V. De Ledinghen1 et A. Villate2
1 Service d’hépatologie, CHU Bordeaux
2 Service hématologie, CHU Tours
Relecteurs :
M. Robin3, D. Valla4, S. Hillaire5, D. Dutheil6, C. Bureau7, A. Plessier4
3 Service d’hématologie, APHP Hôpital Saint-Louis
4 Service d’hépatologie, APHP Hôpital Beaujon et centre de référence des maladies vasculaires du foie
5 Service de gastro-entérologie hépatologie, Hôpital Foch, Paris
6 Association des malades des vaisseaux du foie (AMVF)
7 Service d’hépato-gastroentérologie, CHU Toulouse
Introduction
Le syndrome d’obstruction sinusoïdale (SOS), autrefois appelé « maladie veino-occlusive », est la perte de l’intégrité de la paroi sinusoïdale, caractérisée par l’obstruction concentrique, non thrombotique, des sinusoïdes et de la lumière des veinules hépatiques, en l’absence de lésion primitive ou de thrombose des veines hépatiques. Initialement, les cellules endothéliales sinusoïdales altérées se détachent de la paroi (dénudation de l’endothélium) et migrent vers les zones centrales des lobules hépatiques induisant une obstruction congestive sinusoïdale. La nécrose hépatocellulaire centrolobulaire dépend du niveau d’obstruction. Le SOS peut être associé à d’autres lésions comme une fibrose périsinusoïdale centrolobulaire, une péliose ou une hyperplasie nodulaire régénérative (HNR).
Les premiers cas décrits en 1920 en Afrique du Sud étaient secondaires à l’ingestion de plantes contenant des alcaloïdes de la pyrrozilidine (ergot de Seigle). Le SOS est une complication bien connue des transplantations de cellules souches hématopoïétiques (TCSH). De nombreux médicaments et toxiques ont aussi été associés à la survenue d’un SOS comme par exemple les chimiothérapies ou les traitements immunosuppresseurs (tableau 1). Enfin, l’irradiation corporelle totale ou hépatique et la transfusion de plaquettes ABO incompatible sont aussi associées au SOS. La pathogénie du SOS n’est pas connue.
| Facteurs liés à la greffe |
| – Allogreffe plus à risque que autogreffe– Conditionnement myéloablatif ou séquentiel plus à risque que conditionnement atténué– Deuxième greffe– Irradiation corporelle totale à fortes doses– Donneur haplo-identique– Réactivation du cytomégalovirus (CMV) |
| Facteurs liés aux médicaments (non exhaustif) |
| – Alcaloïdes pyrrolizidiniques– Azathioprine– Busulfan oral ou à fortes doses IV– Carmustine– Cytosine arabinoside– Cyclophosphamide– Dacarbazine– Gemtuzumab ozogamicine– Mitomycine– Oxaliplatine– Phytothérapie– Uréthane– Terbinafine– 6-mercaptopurine– 6-thioguanine |
| Facteurs liés au patient et à sa maladie |
| – Patients âgés– Score de Karnofsky < 90 %– Syndrome métabolique– Maladie réfractaire ou en rechute– Thalassémie– Facteurs génétiques (polymorphisme GSTM1, allèle C282Y, haplotype MTHFR) |
| Facteurs hépatiques |
| – Transaminases pré-greffe > 2,5 N– Bilirubine totale pré-greffe > 1,5 N– Cirrhose– Hépatite virale active– Irradiation hépatique– Surcharge martiale |
Tableau 1
Principaux facteurs de risque de SOS.
L’incidence du SOS est très variable selon les études et diffère selon les facteurs de risque des patients, les médicaments, le nombre de cures de chimiothérapie, le type de conditionnement pré-greffe. L’incidence du SOS est estimée à 14 % après transplantation de moelle ou d’organe, mais avec de grandes différences selon les études et le conditionnement utilisé (0 % à 50 %).
Diagnostic
Manifestations
Les symptômes majeurs du SOS sont non spécifiques et comprennent la prise de poids (rétention hydro-sodée) avec ou sans ascite, l’hépatomégalie, l’hépatalgie et/ou l’ictère. Cependant, la présentation clinique peut être aussi bien peu symptomatique que conduire à une défaillance multiviscérale pouvant entraîner le décès du patient. Après TCSH, le SOS survient généralement durant les trois à quatre premières semaines post-greffe.
On décrit trois formes cliniques de SOS [1] :
- 1)Forme minime : forme limitée ne nécessitant aucun traitement.
- 2)Forme modérée : forme résolutive grâce aux diurétiques.
- 3)Forme sévère : forme ne régressant pas après 100 jours malgré le traitement et pouvant conduire au décès du patient. Les facteurs de mauvais pronostic sont l’élévation du taux de bilirubine, la prise de poids, la cytolyse, l’élévation du gradient de pression hépatique, l’insuffisance rénale et la défaillance multiviscérale.
La mortalité à 100 jours est de 9 % pour les formes minimes et de 100 % pour les formes sévères. La sévérité peut être difficile à appréhender initialement. L’évolution est souvent évocatrice de la gravité lorsque 1) le SOS apparaît rapidement après la greffe, 2) qu’il existe une aggravation rapide devant notamment un temps de doublement de la bilirubine < 24 h, 3) qu’apparait une défaillance multiviscérale.
SOS à l’oxaliplatine
Le risque opératoire a été évalué dans plusieurs études après chimiothérapie à l’oxaliplatine, en particulier pour les métastases hépatiques de cancer du côlon. Dans ce contexte, le SOS est associé à une diminution de la détection des métastases hépatiques, un risque hémorragique opératoire plus élevé, une insuffisance hépatique post-opératoire, ou un retard à la régénération hépatique [2]. L’évolution à long terme du SOS dans ce contexte, après l’arrêt de la chimiothérapie n’est pas bien connue. Des résultats récents montrent que 10 % des patients traités par oxaliplatine, avaient à 6 mois, après en moyenne 5 cycles, une augmentation significative du volume de la rate et du diamètre de la veine porte, en faveur d’une hypertension portale, et 1 % avaient des varices oesophagiennes ou gastriques [3].
Les outils diagnostiques
L’absence d’outils diagnostiques spécifiques peut rendre l’identification d’un SOS difficile. Sa suspicion clinique reste basée sur les critères composites rétrospectifs de Seattle modifié [4] et de Baltimore [5]. En 2016, l’EBMT (European society for Blood and Marrow Transplantation) a proposé de nouveaux critères diagnostiques (tableau 2) en proposant notamment d’inclure la définition de SOS retardé au-delà du 21e jour post-greffe. L’élévation du taux de bilirubine est un marqueur sensible mais peu spécifique de SOS. D’autres marqueurs biologiques ont été proposés mais n’ont pas été validés (procollagène sérique, thrombopénie < 167 G/L, élévation du score APRI ou FIB-4). Les diagnostics différentiels à rechercher et corriger sont une infection, la toxicité hépatique d’un médicament (ciclosporine, azolés, bactrim, autres), les autres causes d’épanchements comme l’insuffisance cardiaque, la pancréatite, les infiltrations hépatiques, les hépatopathies virales (en particulier l’hépatite E…).
| SOS classiquedans les 21 premiers jours après TCSH | SOS retardé> 21 jours après TSCH |
| Bilirubine ≥ 34 μmol/L ET la présence de deux des critères suivants :– Hépatomégalie douloureuse– Gain pondéral > 5 %– Ascite | Critères du SOS classique au-delà du 21e jourOuSOS prouvé histologiquementOuLa présence de deux des critères suivants :– Bilirubine ≥ 34 μmol/L– Hépatomégalie douloureuse– Gain pondéral > 5 %– AsciteETArguments hémodynamiques ou échographiques de SOS |
EBMT : European Society for Blood and Marrow Transplantation ; SOS : syndrome d’obstruction sinusoïdale ; TCSH : transplantation de cellules souches hématopoïétiques.
Tableau 2
Nouveaux critères EBMT pour le diagnostic du SOS chez l’adulte.
L’échographie couplée au doppler hépatique peut être utile pour mettre en évidence une hypertension portale en lien avec le SOS et éliminer les diagnostics différentiels [6]. Les arguments en faveur du SOS comprennent : hépatosplénomégalie, ascite, reperméabilisation de la veine para-ombilicale, épaississement de la paroi de la vésicule biliaire, modification de l’index de résistance de l’artère hépatique, démodulation du flux dans les veines hépatiques, inversion du flux portal. Il est également important de rechercher des arguments pour des diagnostics différentiels comme une insuffisance cardiaque droite, une cirrhose, une thrombose, un obstacle sur les voies biliaires, des lésions infiltrantes du foie. Une échographie-doppler normale n’exclut pas le diagnostic de SOS.
L’imagerie par résonance magnétique (IRM) et le scanner ne sont pas recommandés, ils peuvent cependant parfois être utiles pour rechercher d’autres diagnostics différentiels notamment comme par exemple des foyers profonds de cause infectieuse lorsque l’échographie n’est pas contributive. L’injection de produit de contraste permet alors de voir des troubles de perfusion, avec une image assez caractéristique mais non spécifique, de foie en mosaïque, souvent hypodense sans injection, hyperdense à la phase artérielle et tardive [7].
La ponction biopsie hépatique par voie transjugulaire associée à la mesure du gradient veineux hépatique est l’élément clef du diagnostic de SOS. Elle est souvent préférée à l’abord transcapsulaire compte tenu de la thrombopénie et/ou de l’ascite : elle permet la mesure hémodynamique du gradient de pression hépatique. Elle est cependant rarement réalisée, par crainte de complications hémorragiques et difficultés pratiques. Après transplantation de moelle, le gradient veineux hépatique > 10 mmHg a une sensibilité de 52 %, une spécificité de 91 % et une valeur prédictive positive > 85 % pour le diagnostic de SOS. Une étude rétrospective récente a analysé les résultats de 54 biopsies (dont 47 par voie transjugulaire) chez 45 patients après TCSH. Aucun effet secondaire de la biopsie n’est rapporté. Il y avait 5 % de SOS et 30 % de GVH (réaction du greffon contre l’hôte). L’association entre les symptômes cliniques, biologiques, et de l’imagerie et le diagnostic final de la biopsie étaient mauvais. Seuls 34 % des hypothèses avant biopsie étaient confirmées par la biopsie [8]. Une étude ancienne renforce ces résultats et montre que la biopsie hépatique ne confirme le diagnostic évoqué de SOS que dans 58 % des cas, et à l’inverse qu’un SOS est présent dans 6 % des cas alors que le diagnostic n’était pas évoqué [9].
Les premières lésions sont présentes 6 à 8 jours après l’exposition à l’agent toxique. Il existe un élargissement oedémateux de l’espace sous-endothéliale, contenant des débris cellulaires et des erythrocytes fragmentés, entre la membrane basale et l’adventice des veines centro et médiolobulaires. Les sinusoïdes sont congestifs et dilatés, et la nécrose hépatocytaire centrolobulaire associée sont souvent plus étendues que l’atteinte de la veinule. Plus tardivement apparaît une fibrose autour des sinusoïdes puis un épaississement et une occlusion des veinules centrolobulaires. Au cours des SOS liés à l’oxaliplatine, la prévalence de cette occlusion est de 48% [10].
La sévérité clinique du SOS est proportionnelle à la présence, à l’extension et à l’association :
- –de la nécrose hépatocytaire,
- –de la fibrose sinusoïdale,
- –de l’épaississement excentré de l’espace sous-endothéliale de la veinule,
- –de la phlébosclérose,
- –de l’obstruction veinulaire hépatique [11].
La plupart des études après TCSH décrivent une occlusion des veines centro-lobulaires observée chez 50 % des patients avec SOS minime ou modéré et 75 % des patients avec SOS sévère.
Enfin, plus tardivement une hyperplasie nodulaire régénérative est possible. L’absence de signes histologiques spécifiques de SOS n’exclut pas le diagnostic. Les anomalies histologiques peuvent être hétérogènes en fonction du site de ponction. Ces résultats méritent d’être confirmés et corrélés à l’évolution clinique car les critères diagnostiques de SOS dans ces études sont souvent des critères cliniques et non histologiques.
Prévention et traitement du SOS
La prévention du SOS
La recherche de facteurs de risque est essentielle pour la prévention du SOS.
Les facteurs de risque de SOS sont une maladie hépatique préexistante, un antécédent de SOS, un antécédent d’irradiation abdominale, de traitement par Mylotarg®, une deuxième greffe, un âge adulte ou < 2 ans et en pédiatrie : lymphohistiocytose familiale et autres syndromes d’activation macrophagique, ostéopétrose, leucodystrophie métachromatique. En cas de tumeur solide (en particulier les métastases hépatiques d’un cancer colorectal), une élévation de la gamma glutamyl-peptidase avant chirurgie, l’âge, le sexe féminin, le taux de rétention du vert d’indocyanine à 15 minutes, le nombre de cycles de chimiothérapie, et une période courte entre la fin de la chimiothérapie et la chirurgie digestive sont autant de facteurs de risque de SOS. Le meilleur traitement préventif du SOS dans ces situations est de réduire au maximum l’intensité de la chimiothérapie et de la radiothérapie. D’autres mesures peuvent être instituées avant la greffe ou les traitements toxiques pour les sinusoïdes, comme la chélation du fer en cas de surcharge martiale, les traitements antiviraux de l’hépatite B et de l’hépatite C réplicative.
Les traitements du SOS
La prophylaxie par héparine n’a pas de bénéfice sur le SOS [12].
L’acide ursodésoxycholique en prophylaxie primaire jusqu’à J90 post-TCSH permet une diminution de la mortalité non liée à la rechute et une augmentation de la survie globale [13]. L’étude ne montre pas clairement une diminution de l’incidence de SOS mais une baisse du nombre de patients présentant une bilirubine élevée.
Le défibrotide (oligodésoxyribonucléotides simple brin extraits d’ADN de la muqueuse intestinale de porc) a de multiples propriétés antithrombotiques, fibronolytiques et angiogéniques. Son mécanisme d’action dans le SOS n’est pas complètement élucidé [14]. Son intérêt pour la prophylaxie primaire n’a été démontré que chez l’enfant dans un essai contrôlé randomisé de phase III après TCSH en présence de facteurs de haut risque [15].
En traitement curatif, le défibrotide (posologie : 25 mg/kg/j en 4 injections) a été évalué chez l’adulte présentant un SOS avec défaillance multiviscérale au cours d’une phase III contrôlée non randomisée comparant 102 patients traités par défibrotide de 2006 à 2008 versus 32 patients non traités d’un groupe historique de 1995 à 2007. L’analyse statistique soulignait un avantage en survie à J100 post-greffe en faveur du défibrotide (32 % versus 25 %, p < 0,01) sans données après cette période. L’autorisation de mise sur le marché (AMM) a été obtenue en 2014 (service médical rendu modéré, ASMR IV) dans les formes sévères à la posologie de 25 mg/kg/j en 4 injections pendant au moins 21 jours, éventuellement raccourci à 14 jours en cas de résolution rapide des symptômes [16]. Son utilisation dans les SOS modérés reste encore discutée étant donné le risque d’évolution rapide vers une forme sévère et la possibilité de réduire la mortalité à J100 post-greffe en cas d’initiation précoce du traitement [17, 18]. Les recommandations de la société britannique de greffe de moelle (BSBMT) réserve l’utilisation du défibrotide dans les SOS modérés en cas de situations à risque [19] (hépatopathie pré-existante, seconde allogreffe, conditionnement myélo-ablatif comprenant du busulfan, utilisation en pré-greffe de gemtuzumab-ozogamicin).
Le risque hémorragique sous défibrotide est estimé à 15 % chez les 419 patients des quatre études réalisées au cours de son développement. La commission de transparence de la Haute Autorité de Santé (HAS) recommande de maintenir si possible un taux de plaquettes entre 35-50 G/L en cas de traitement curatif. Enfin, le défibrotide reste un médicament orphelin dans cette indication dont l’impact pharmaco-économique reste à déterminer.
Le traitement du SOS repose sur des soins de support des symptômes : traitement de la rétention hydrosodée par des diurétiques, maintien d’une pression artérielle suffisante (transfusion et albumine en cas d’anasarque), arrêt des lipides en cas d’alimentation parentérale, limiter les hépatotoxiques. Le shunt porto-systémique intrahépatique (TIPS), les dérivations chirurgicales ou la transplantation hépatique ont été décrits dans quelques cas. La transplantation hépatique peut être discutée en cas de SOS sévère en fonction du pronostic de la pathologie hématologique ou en cas de SOS sévère toxique (intoxication aux alcaloïdes de la pyrozilidine).
Recommandations
- •Rechercher un SOS en cas d’anomalie du bilan sanguin hépatique après transplantation de cellules souches hématopoïétiques, chimiothérapie, traitement immunosuppresseur après transplantation d’organe, ou maladie inflammatoire de l’intestin (B).
- •Rechercher un SOS en cas de prise de poids, avec ou sans ascite, hépatomégalie et ictère après exclusion d’autres causes (sepsis, toxicités médicamenteuses, GVH) (C).
- •En cas de suspicion diagnostique, réaliser une ponction-biopsie hépatique par voie trans-jugulaire avec évaluation hémodynamique (C).
- •Assurer le monitorage des facteurs de risque de SOS (B).
- •Utiliser l’acide ursodésoxycholique en prophylaxie primaire dans les greffes de cellules souches hématopoïétiques jusqu’à J90 (B).
- •Utiliser le défibrotide en traitement curatif du SOS dans les formes sévères chez les patients adultes (C) et au cas par cas dans les formes modérées chez les patients à haut risque de SOS, si possible authentifié par une histologie, au cours des transplantations de cellules souches hématopoïétiques (C).
- •Informer les patients de l’existence des associations de patients dès l’annonce du diagnostic.
Références
1. Mohty M, Malard F, Abecassis M, Aerts E, Alaskar AS, Aljurf M, et al. Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients : a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant 2016 ; 51 : 906-912.
2. Rubbia-Brandt L, Lauwers GY, Wang H, Majno PE, Tanabe K, Zhu AX, et al. Sinusoidal obstruction syndrome and nodular regenerative hyperplasia are frequent oxaliplatin-associated liver lesions and partially prevented by bevacizumab in patients with hepatic colorectal metastasis. Histopathology 2010 ; 56 : 430-9.
3. Gioia S. Non cirrhotic portal hypertension secondary to oxaliplatin therapy : Incidence and presentation. J Hepatol 2018 ; 68 (1) : 701.
4. Shulman HM, Hinterberger W. Hepatic veno-occlusive disease--liver toxicity syndrome after bone marrow transplantation. Bone Marrow Transplant. 1992 ; 10 : 197-214.
5. Jones RJ, Lee KS, Beschorner WE, Vogel VG, Grochow LB, Braine HG, et al. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation. 1987 ; 44 : 778-83.
6. Lassau N, Leclère J, Auperin A, Bourhis JH, Hartmann O, Valteau-Couanet D, et al. Hepatic veno-occlusive disease after myeloablative treatment and bone marrow transplantation : value of gray-scale and Doppler US in 100 patients. Radiology. 1997 ; 204 : 545-552.
7. Chiou SY, Lee RC, Chi KH, Chia-Hsien Cheng J, Chiang JH, et al. The triple-phase CT image appearance of post-irradiated livers. Acta Radiol 2001 ; 42 (5) : 526-31.
8. Michonneau D, Ruggiu M, Bedossa P, et al. Interest of liver biopsy for patients with misunderstood liver blood test anomalies after allogeneic hematopoietic stem cell transplantation. Biology of Blood and Marrow Transplantation. In press.
9. Carreras E, Grañena A, Navasa M, et al. Transjugular liver biopsy in BMT. Bone Marrow Transplant 1993 ; 11 (1) : 21-6.
10. Rubbia-Brandt L, Audard V, Sartoretti P, et al. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann Oncol 2004 ; 15 (3) : 460-6.
11. Shulman HM, Fisher LB, Schoch HG, Henne KW, McDonald GB. Veno-occlusive disease of the liver after marrow transplantation : histological correlates of clinical signs and symptoms.Hepatology 1994 ; 19 (5) : 1171-81.
12. Imran H, Tleyjeh IM, Zirakzadeh A, Rodriguez V, Khan SP. Use of prophylactic anticoagulation and the risk of hepatic veno-occlusive disease in patients undergoing hematopoietic stem cell transplantation : a systematic review and meta-analysis. Bone Marrow Transplant 2006 ; 37 : 677-686.
13. Ruutu T, Juvonen E, Remberger M, Remes K, Volin L, Mattsson J, et al. Improved Survival with Ursodeoxycholic Acid Prophylaxis in Allogeneic Stem Cell Transplantation : Long-Term Follow-Up of a Randomized Study. Biol. Blood Marrow Transplant 2014 ; 20 : 135-138.
14. Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood 2016 ; 127 (13) : 1719-27.
15. Corbacioglu S, Cesaro S, Faraci M, Valteau-Couanet D, Gruhn B, Rovelli A, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation : an open-label, phase 3, randomised controlled trial. Lancet 2012 ; 379 : 1301-1309.
16. Richardson PG, Riches ML, Kernan NA, Brochstein JA, Mineishi S, Termuhlen AM, et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 2016 : 127 : 1656-1665.
17. Bazarbachi RA. La Maladie Veino-occlusive – syndrome d’obstruction sinusoidale Mise à jour 2014. Hematologie 2015 ; 21 (3) : 35-41.
18. Richardson PG, Smith AR, Triplett BM, Kernan NA, Grupp SA, Antin JH, et al. Earlier defibrotide initiation post-diagnosis of veno-occlusive disease/sinusoidal obstruction syndrome improves Day +100 survival following haematopoietic stem cell transplantation. Br J Haematol 2017 ; 178 : 112-118.
19. Dignan FL, Wynn RF, Hadzic N, Karani J, Quaglia A, Pagliuca A, et al. BCSH/BSBMT guideline : diagnosis and management of veno-occlusive disease (sinusoidal obstruction syndrome) following haematopoietic stem cell transplantation. Br J Haematol 2013 ; 163 : 444-457.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International