Hépato-Gastro & Oncologie Digestive
MENULe syndrome LPAC ou lithiase biliaire avec phospholipides bas Volume 23, numéro 5, Mai 2016

1422, route des Mauvares,
13840 Rognes,
France
- Mots-clés : lithiase cholestérolique, MDR3, ABCB4, acide ursodesoxycholique, cholestérol, phospholipides, acides biliaires, lithiase intrahépatique, cirrhose biliaire, cholangiocarcinome
- DOI : 10.1684/hpg.2016.1295
- Page(s) : 377-83
- Année de parution : 2016
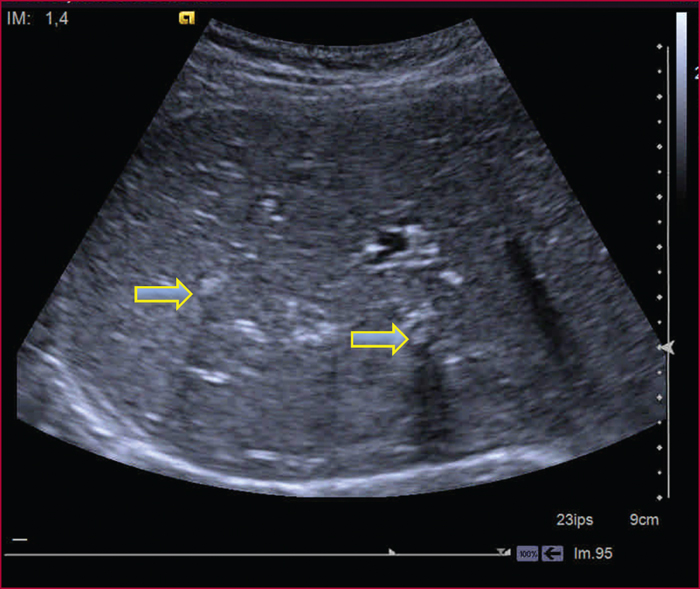
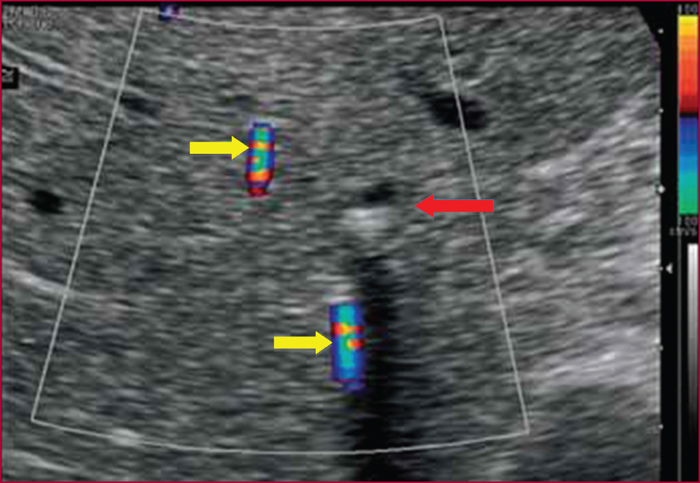
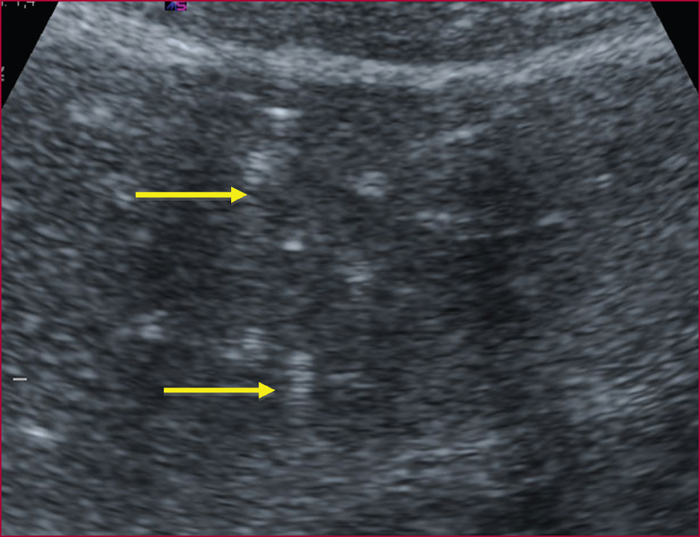
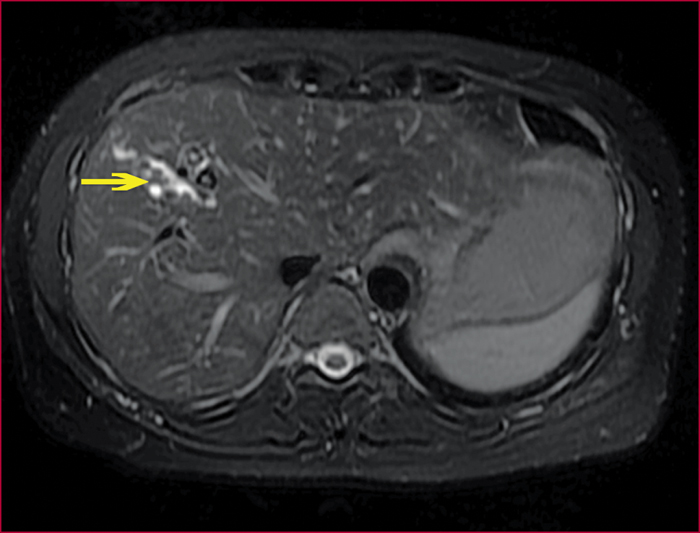
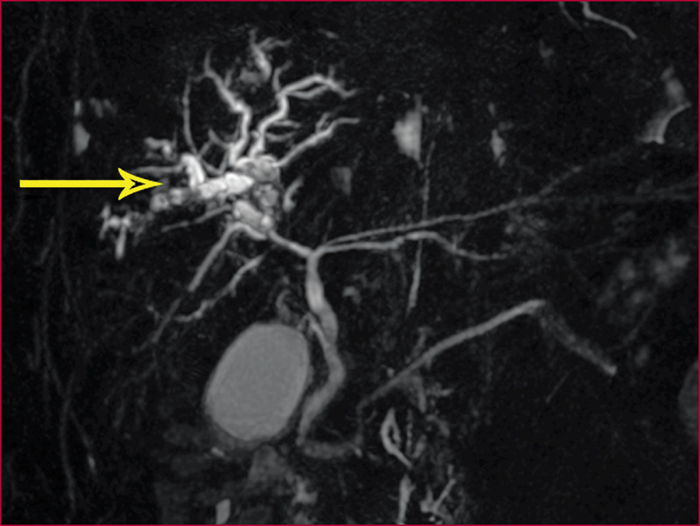
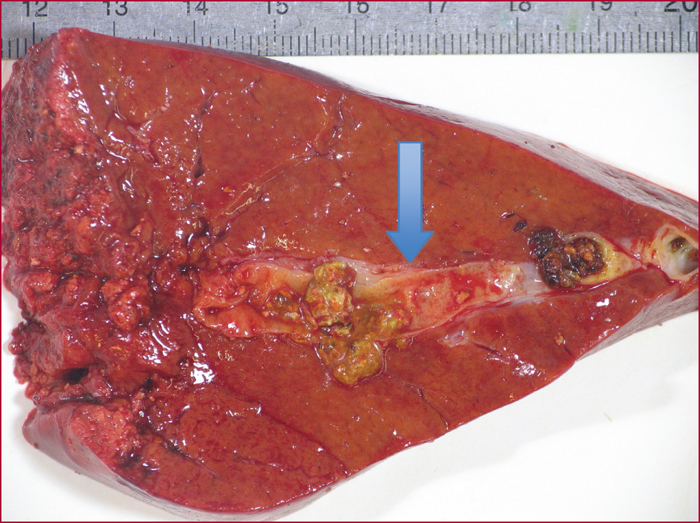
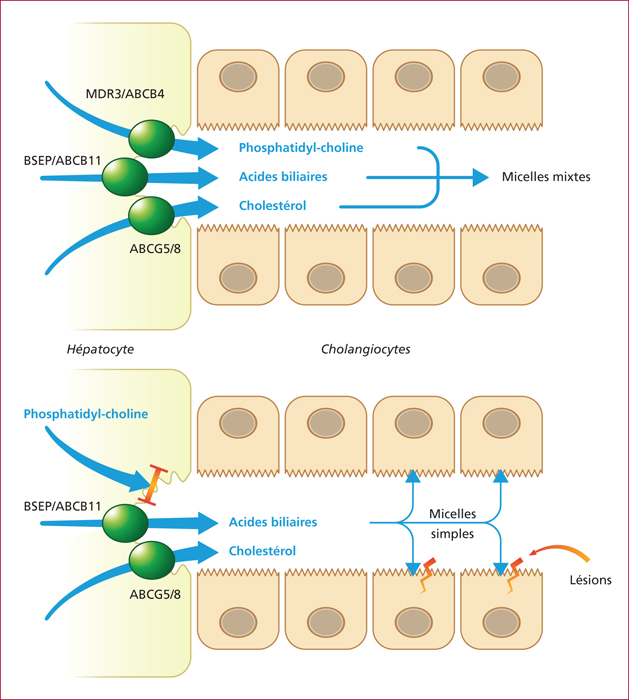
Le syndrome LPAC (Low phospholipid-associated cholelithiasis) est une maladie génétique caractérisée par une lithiase cholestérolique vésiculaire et/ou intrahépatique. Elle est causée par une mutation du gène ABCB4 qui code la protéine du canalicule biliaire MDR3, une floppase qui joue un rôle crucial dans la sécrétion de la phosphatidyl choline dans la bile. Le diagnostic doit être suspecté en présence des manifestations suivantes : 1) Symptômes biliaires (douleur biliaire, angiocholite, ictère, pancréatite aiguë) survenant avant 40 ans. 2) Récidive des symptômes biliaires après cholécystectomie. 3) Antécédents de cholestase gravidique. 4) Histoire familiale de lithiase chez les apparentés du premier degré. 5) Présence d’images échogènes intrahépatiques, de « queue de comète », ou d’un artéfact de scintillement en échographie Doppler couleur, traduisant la présence de calculs ou de boue biliaire dans le foie. Dans les cas douteux, le diagnostic peut être confirmé par une recherche de la mutation. Le traitement par l’acide ursodesoxycholique entraîne une amélioration rapide, spectaculaire et durable des symptômes dans la majorité des cas et il prévient les complications. Dans quelques cas cependant, la chirurgie peut être nécessaire.