Hématologie
MENU06 Leucémies aiguës (biologie et clinique) Volume 25, supplément 1, Mars 2019

06-01
Évaluation du protocole FLAG dans les leucémies aiguës myéloblastiques réfractaires et en rechute au service d’hématologie de Sidi Bel Abbès, Algérie
N. Siali*1, M. Benlazar1, S. Benichou2, A. Hadjeb1, A. Elmestari3, K. Tayebi4, N. Zemri1, Z. Z. zouaoui5
1 Hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Algérie ; 2 hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Sidi Bel Abbès, Algérie, Sidi Bel Abbès, Algérie ; 3 Hématologie, chu, Sidi Bel Abbès, Algérie ; 4 Service d’hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Algérie ; 5 Hématologie, CHU Bel Abess, Sidi Bel Abbès, Algérie
Introduction. La leucémie aiguë myéloblastique (LAM) est une affection redoutable, la chimiothérapie intensive permet d’obtenir des rémissions complètes RC cependant avec des rechutes essentiellement dans les deux premières années, la greffe de moelle osseuse allogénique reste la seule alternative thérapeutique sous réserve d’obtenir une RC par des protocoles agressifs.
Patients et méthodes. Notre étude est rétrospective analytique entre 2007 et 2017 prenant en compte les caractères cliniques et thérapeutiques des malades réfractaires donc ne répondant pas à deux cycles d’induction par le protocole (3+7), et en rechute précoce (avant 12 mois de RC) et tardives après 12 mois, traités par le protocole FLAG (fludarabine 30 mg/m2 pendant 5 j et cytarabine 2 g/m2 pendant 5 j et Granocyte à compter du 1̊ j et jusqu’a la sortie d’aplasie la survie est calculée selon la méthode de Kaplan-Meier.
Résultats. Nous avons colligé 50 patients dont 26 hommes et 24 femmes avec un sex-ratio de 1,1, l’âge médian est de 35 ans avec des extrêmes de 15 à 60 ans, 22 patients étaient réfractaires et 28 ont rechuté de façon précoce (15 cas), ou tardive (13 cas), le délai moyen de la rechute est de 6 mois avec des extrêmes de 2 à 60 mois la RC est obtenue dans 38 % (19 cas), échec dans 32 % (16 cas), décès 30 % (15 cas), la tolérance au traitement était bonne l’aplasie postchimiothérapie a duré en moyenne 10 j (7-20 j), grade II et III, la fièvre est l’événement indésirable le plus fréquent liée aux infections bactériennes et fongiques cependant bien contrôlées n = 15 (83,3%), la survenue d’un syndrome hémorragique lié à la thrombopénie n = 10 (55,5%), des troubles métaboliques ont été observés dans 44,4 % des cas 4 patients ont bénéficié d’une greffe de moelle osseuse allogénique, la médiane de survie est 5 mois avec des extrêmes de 1 mois à 9 ans.
Conclusion. La LAM réfractaire et en rechute est de mauvais pronostic, le protocole FLAG semble efficace et bien toléré il peut induire des rémissions qui permettent de réaliser des greffes allogéniques, il est important de bien définir le profil biomoléculaire et cytogénétiques afin de mieux stratifier le traitement de rattrapage.
06-02
Prise en charge des rechutes des leucémies aiguës myéloblastiques au service d’hématologie de Sidi Bel Abbès, Algérie
N. Siali*1, M. Benlazar1, S. Benichou2, A. Hadjeb1, F. Ouaddah1, Z. Z. zouaoui3
1 Hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Algérie ; 2 hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Sidi Bel Abbès, Algérie, Sidi Bel Abbès, Algérie ; 3 Hématologie, CHU Bel Abess, Sidi Bel Abbès, Algérie
Introduction. Les leucémies aiguës myéloblastiques (LAM) sont les plus graves d’hémopathies malignes dues à la prolifération blastique médullaire et périphérique et se manifestant par des signes d’insuffisance sanguine avec ou sans syndrome tumoral, la chimiothérapie permet d’obtenir une rémission tandis que la greffe de moelle osseuse allogénique reste le seul traitement curateur.
Patients et méthodes. Notre étude est analytique et rétrospective prenant en compte les caractères cliniques et thérapeutiques des patients entre 2007 et 2017, nous avons colligé 28 cas de rechutes précoces survenant avant 12 mois après obtention de la rémission complète RC et tardives après les 12 mois, les malades ont été traités selon deux protocoles de rattrapage FLAG (fludarabine 30 mg/m2 pdt 5 J et cytarabine 2 g/m2 pdt 5 J et Granocyte à compter du premier jour et jusqu’à la sortie de l’aplasie) et HAMAC (cytarabine 200 mg/m2 pdt 3 j et amsacrine 100 mg/m2 pdt 3 j), la survie est calculée selon la méthode de Kaplan et Meier.
Résultats. Au total 28 cas de rechutes ont été analysés parmi 90 cas de LAM et parmi ces rechutes nous avons 15 cas de rechutes précoces et 13 cas de rechutes tardives, nous avons compté 13 femmes et 15 hommes donnant un sex-ratio de 1,1, l’âge médian est de 45 ans avec des extrêmes de 17 à 66 ans, pour les comorbidités : 48 % ont un diabete, 22 % ont HTA, et 30 % n’ont aucune tare, tous les malades ont reçu un à deux cycles d’induction par le protocole (3+7), le délai médian de la rechute est de 6 mois avec des extrêmes de 2 à 60 mois 25 patients ont reçu le FLAG et 3 ont reçu HAMAC, nous avons obtenu une RC dans 33 % (9 cas), échec 30 % (8 cas), décès 37 % (11 cas), les causes de décès : syndrome hémorragique sévère 40 % (10 cas), choc septique 48 % (12 cas), localisation extramédullaire 8 % (2 cas), cardiopathie 4 % (1 cas), 4 patients ont été greffés, la médiane de survie est de 5 mois avec des extrêmes de 1 mois à 9 ans.
Conclusion. Les LAM restent à ce jour de mauvais pronostic, la greffe de moelle osseuse allogénique est le seul traitement curateur quand il existe un donneur HLA compatible et en dehors des facteurs de mauvais pronostic essentiellement génétiques et biomoléculaires.
06-03
Impact de la signalisation du récepteur de la cellule T dans la leucémogenèse des thymocytes déficients en Pten
M. Loosveld*1, S. Gon2, D. Potier2, G. Michel3, N. Vey4, B. Malissen2, R. Roncagalli2, B. Nadel2, D. Payet-Bornet2
1 Laboratoire d’hématologie, Hôpital de la Timone ; Aix Marseille Université CIML, Marseille ; 2 CIML, Aix-Marseille Université, Marseille ; 3 Service d’hématologie et oncologie pédiatrique, Hôpital de la Timone, Marseille ; 4 Epigenetic factors in normal and malignant hematopoiesis, CRCM-Inserm, U1068 ; Institut Paoli-Calmettes ; Aix-Marseille Université, UM 105 ; CNRS, UMR7258, Marseille
Introduction. Au cours de la différentiation thymique ab, la qualité du récepteur de la cellule T (TCR) est testée et détermine le destin du thymocyte. Ce contrôle qualité du TCR est réalisé par deux processus : la sélection positive et la sélection négative. Au cours de la sélection positive, les cellules exprimant un TCRab ‘fit’ c’est-à-dire reconnaissant avec une affinité suffisante le CMH classe I ou classe II sont sélectionnées et deviendront des LT CD8+ ou CD4+ respectivement. Si le TCR s’avère incapable de reconnaitre le CMH (i.e. le TCR est unfit), les thymocytes seront négligés et mourront. Le but de la sélection négative est d’éliminer les lymphocytes T (LT) potentiellement autoréactifs. De ce fait, si le TCRαβ reconnaît avec une forte affinité les complexes CMH-peptides de soi, les LT sont éliminés par apoptose. Ainsi, au cours de la thymopoïèse, la signalisation TCR peut induire deux effets biologiques opposés qui sont l’apoptose et la survie/prolifération cellulaire. Récemment, nous avons cherché à comprendre comment cette signalisation TCRab s’intègre dans la leucémogenèse des thymocytes.
Matériels et méthodes. La majorité des leucémies aiguës lymphoblastiques T (LAL-T) matures TCRab+ présente une perte de fonction du gène suppresseur de tumeur PTEN. Ainsi dans cette étude, les thymocytes murins Pten-déficients (dénommés Ptendel) ont été utilisés comme modèle de leucémie.
Résultats. Nos travaux révèlent que la signalisation TCRab est activement impliquée dans la leucémogenèse médiée par l’inactivation de PTEN. En effet, nos résultats suggèrent que les thymocytes Ptendel qui possèdent un TCR fit sont éliminés et ne peuvent pas développer de leucémies. En revanche, les thymocytes Ptendel ne possédant pas de TCR ou ayant un TCR unfit peuvent se transformer en T-LBL (lymphome lymphoblastique T) ou en LAL-T respectivement.
Conclusion. En conclusion, dans un contexte PTEN-déficient, une signalisation TCRab correcte empêche le développement leucémique et agit donc comme un suppresseur de tumeur. De plus, cette observation suggère que la perte de Pten qui est généralement associée à la survie/prolifération cellulaire, peut dans certaines conditions induire l’apoptose.
06-04
Efficacité et tolérance du quizartinib, inhibiteur puissant et sélectif de FLT3, en monothérapie chez des patients atteints d’une leucémie aiguë myéloïde en rechute/réfractaire avec mutation FLT3-ITD inclus dans l’essai de phase III international, randomisé et contrôlé QuANTUM-R
H. Dombret*1, J. Cortes2, S. Khaled3, G. Martinelli4, A. Perl5, S. Ganguly6, N. Russel7, A. Kramer8, D. Hogge9, B. Jonas10, A. Leung11, P. Mehta12, P. Montesinos13, M. Radsak14, S. Sica15, M. Arunachalam16, M. Holmes16, K. Kobayashi16, R. Namuyinga16, N. Ge16, A. Yver16, Y. Zhang16, M. Levis17
1 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 2 Hématologie, MD Anderson Cancer Center, Texas City, États-Unis ; 3 Department of hematology/hct, City of Hope national Medical Center, Duarte, États-Unis ; 4 Irst, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori IRCCS, Meldola, Italie ; 5 Division of hematology-oncology, Université de Pennsylvanie, Philadelphie, États-Unis ; 6 Department of hematology, University of Kansas Medical Center, Kansas City, États-Unis ; 7 Department of hematology, Université de Nottingham, Nottingham, Royaume Uni ; 8 Klinische kooperationseinheit molekulare hämatologie/onkologie, Universität Heidelberg and German Cancer Research Center, Heidelberg, Allemagne ; 9 Department of hematology, Vancouver General Hospital, Vancouver, Canada ; 10 Department of hematology, UC Davis Comprehensive Cancer Center, Sacramento, États-Unis ; 11 Department of hematology, University of Hong Kong, Hong Kong, Chine ; 12 Department of hematology, Université de Bristol, Bristol, Royaume Uni ; 13 Department of hematology, Hospital Universitari i Politècnic La Fe, València, Espagne ; 14 Department of hematology, medical oncology and pneumology, University Medical Center of the Johannes Gutenberg University, Mayence, Allemagne ; 15 Hematology department, Fondazione Policlinico Universitario A. Gemelli IRCCS - Università Cattolica del Sacro Cuore, Rome, Italie ; 16 Dsi, Daiichi Sankyo, Inc., Basking Ridge, États-Unis ; 17 Department of hematology, The Sidney Kimmel Comprehensive Cancer Center, Baltimore, États-Unis
Introduction. Les mutations FLT3-ITD surviennent chez environ 25 % des patients (pts) atteints de leucémie aiguë myéloïde (LAM) et sont associées à une hyperleucocytose, un risque élevé de rechute, une moins bonne réponse aux traitements de rattrapage et une survie globale (SG) plus courte. Les pts atteints de LAM FLT3-ITD en rechute ou réfractaire (R/R) ont un moins bon pronostic et représentent un important besoin médical non satisfait. Quizartinib (Q) est un inhibiteur de FLT3 (FLT3i) puissant et sélectif administré par voie orale une fois par jour. QuANTUM-R est le premier essai international de phase III, randomisé et contrôlé (NCT02039726), à démontrer qu’un FLT3i en monothérapie améliorait la SG comparativement à une chimiothérapie (CT) chez des pts atteints d’une LAM FLT3-ITD R/R.
Patients et méthodes. Des pts adultes atteints d’une LAM FLT3-ITD R/R (première rémission ≤ 6 mois) après un traitement standard de LAM (± greffe) ont été randomisés 2:1 entre Q (53 mg, phase d’induction à 26,5 mg) et CT au choix de l’investigateur (LoDAC, MEC, FLAG-IDA). Les pts recevant une greffe dans le bras Q pouvaient reprendre le traitement après la greffe. Jusqu’à 2 cycles par MEC ou FLAG-IDA étaient autorisés alors que Q ou LoDAC étaient administrés tant qu’un bénéfice était observé ou jusqu’à survenue d’une toxicité inacceptable. Les critères d’évaluation primaires et secondaires étaient la SG et la survie sans événement (SSE). Des analyses de sensibilité pour la SG et la SSE et des analyses de sous-groupes de la SG ont été effectuées. Les critères d’évaluation exploratoires comprenaient : taux et durée de réponse, taux de greffes et effets indésirables apparus sous traitement (EIAT).
Résultats. 367 pts ont été randomisés ; 245 dans le bras Q vs 122 dans le bras CT (LoDAC n = 29; MEC n = 40; FLAG-IDA n = 53). Le suivi médian était de 23,5 mois (mo). 6 pts recevaient toujours le traitement initial dans le bras Q à la date d’arrêté des données contre 0 dans le bras CT. Les caractéristiques à l’inclusion, telles que l’âge, la réponse au traitement antérieur, les antécédents de greffe ou le ratio allélique FLT3-ITD étaient équilibrées entre les deux bras. Le risque relatif (RR) de la SG pour Q par rapport à la CT était de 0,76 (p = 0,0177) avec une SG médiane de 6,2 mo vs 4,7 mo et une SG estimée à 12 mo de 27 % vs 20 % dans les bras Q et CT respectivement. Le RR de la SSE était de 0,90 (p = 0,1071); la SSE médiane était de 1,4 mo vs 0,9 mo respectivement. Les analyses de sensibilité de la SG (censure pour la greffe, pour utilisation ultérieure de FLT3i ou dans la population per protocole) et de la SSE (population per protocole) et les analyses de sous-groupes de la SG ont confirmé le bénéfice de Q par rapport à la CT. Le taux de réponse complète composite (RCc) était de 48 % vs 27 % et la durée de RCc était de 12,1 vs 5,0 semaines dans les bras Q et CT respectivement. Les taux de greffes étaient de 32 % (Q) et 12 % (CT). Les taux d’EIAT étaient comparables, malgré une durée de traitement plus longue dans le bras Q. Les EIAT de grade ≥ 3 les plus fréquents dans les deux bras étaient les infections et ceux associés à une cytopénie. Seuls 2 pts ont dû interrompre Q pour allongement de l’intervalle QT (grade 2). Le taux de QTcF > 500 ms était de 3 % dans le bras Q. Aucun QTcF de grade 4 n’a été observé.
Conclusion. Ce rapport confirme le gain en SG observé avec Q en monothérapie par rapport à la CT chez des pts atteints d’une LAM FLT3-ITD R/R ainsi qu’un profil de tolérance favorable, démontrant un bénéfice clinique significatif chez des pts qui ont peu d’options et constituant un changement de paradigme dans la prise en charge de ces pts.
06-05
Analyse de la performance de l’alarme « blastes » sur Advia® 2120/2120i. Résultats d’une étude multicentrique
F. Aidoudi1, V. BACCINI*2, B. Bardet3, C. Lafon4, A. Pellicier5, F. Reins6, P. Tales7
1 Laboratoire d’hématologie, CH Geneviève de Gaulle Anthonioz, Saint-Dizier ; 2 Laboratoire d’hématologie, Hôpital Nord, Marseille ; 3 Laboratoire d’hématologie, CH de Bourg-en-Bresse, Bourg-en-Bresse ; 4 Laboratoire d’hématologie, Hôpital Robert Debré, Reims ; 5 Laboratoire d’hématologie, CH de Martigues, Martigues ; 6 Laboratoire d’hématologie, Centre Hospitalier de Saint-Dié-des-Vosges, Saint-Dié-des-Vosges ; 7 Laboratoire d’hématologie, CH de Charleville-Mézières, Charleville-Mézières
Introduction. L’une des principales préoccupations des laboratoires d’hématologie est de détecter le plus tôt possible les hémopathies, ou les rechutes éventuelles. La détection des cellules blastiques dans le sang circulant est donc une des priorités des laboratoires de ville et hospitaliers.
L’objet de cette étude a été de vérifier la concordance de la semi-quantification du pourcentage de blastes, rendue par l’hématimètre ADVIA 2120/2120i, avec le pourcentage de blastes quantifié manuellement par le laboratoire.
Matériels et méthodes. Sept sites hospitaliers ont été inclus dans cette étude prospective, 115 603 échantillons de patients hospitalisés ont été testés entre décembre 2016 et avril 2017. L’alarme Blaste a été déclenchée sur 1559 échantillons (soit 1,35 % de la population étudiée).
Résultats. La sensibilité et la spécificité de l’alarme Blastes sont respectivement de 89,04 % et de 98,87 %. La valeur prédictive négative est de 99,97 %.
De manière générale, le niveau de l’alarme émis par l’hématimètre en nombre de croix (de + à +++), est en accord avec le pourcentage de blastes comptés au microscope.
Conclusion. Le non déclenchement de l’alarme Blastes sur ADVIA 2120/2120i apparaît comme un outil fiable pour attester l’absence de blastes dans un échantillon sanguin.
La reproductibilité de l’alarme Blastes et la bonne cohérence du niveau d’alarme avec le pourcentage observé au microscope, peuvent aider à l’adaptation des recommandations des sociétés savantes, afin d’optimiser le recours au microscope.
06-06
Blinatumomab pour traiter la maladie résiduelle minime chez des adultes atteints de leucémie aiguë lymphoblastique à précurseurs B : médiane de survie non atteinte chez les patients en réponse maladie résiduelle complète après un suivi médian de 53,1 mois
H. Dombret*1, N. Gökbuget2, G. Zugmaier3, M. Bonifacio4, C. Graux5, C. Faul6, MS. Topp7, M. Brüggemann8, K. Taylor9, R. Bargou10
1 Hématologie, chu saint-louis, Paris ; 2 Medizinische klinik ii, Goethe University Hospital, Francfort, Allemagne ; 3 Département médical, Amgen Research (Munich) GmbH, München, Allemagne ; 4 Department of medicine, section of hematology, Université de Vérone, Vérone, Italie ; 5 Service d’hématologie, CHU UCL Mont-godinne-dinant, Dinant, Belgique ; 6 Service de médecine interne, UniversitätsKlinikum Tübingen, Tubingue, Allemagne ; 7 Department of internal medicine ii, Universitätsklinikum Würzburg, Wurtzbourg, Allemagne ; 8 Medizinische klinik und poliklinik, University Schleswig Holstein in the City Hospital, Kiel, Allemagne ; 9 Biostatistics, Amgen, Thousand Oaks, États-Unis ; 10 Comprehensive cancer center mainfranken, Universitätsklinikum Würzburg, Wurtzbourg, Allemagne
Introduction. Environ 30 % des adultes atteints de LAL-B obtenant une rémission hématologique complète (RCh) après chimiothérapie intensive ont une MRD persistante ou récurrente (MRD+), détectée par PCRq en temps réel ou en cytométrie de flux. MRD : prédicteur de rechute le plus fiable dans LAL-B. Blin : anticorps bispécifique qui redirige les lymphocytes T pour induire la destruction de cellules cibles CD19+. Dans BLAST, étude internationale de Phase 2 conduite chez des patients (pts) adultes atteints de LAL-B MRD+, 78% (88/113) des pts évaluables ont obtenu une réponse MRD complète après le C1 de traitement (ttt) par Blin. Incidence des événements indésirables (EI) de grade 3 ou 4, dont événements neurologiques (13 %) et syndromes de libération des cytokines (2 %) similaire à celle observée dans d’autres études testant le blin. Avec un suivi minimal de 18 mois, survie globale (SG) médiane : 36,5 mois (IC95% : 19,8-NE). Nous rapportons ici les résultats après un suivi minimal de 3 ans.
Patients et méthodes. BLAST inclut pts adultes avec LAL-B en première (RC1), deuxième ou plus (RC2+) RCh après ≥ 3 schémas de chimiothérapie intensive, avec MRD+ au seuil de 10-3, 2 semaines au moins après dernière chimiothérapie. Tous les pts ont reçu du blin 15 μg/m2/j pendant 4 cycles maximum (1 cycle : 4 semaines de perfusion continue puis 2 semaines sans ttt). Réponse MRD complète (RMC) : absence d’amplification de la cible avec sensibilité minimale de 10-4. Après évaluation de réponse MRD à la fin du C1, pts peuvent recevoir une greffe de cellules souches hématopoïétiques (SCH). SG estimée par méthode de Kaplan-Meier (globalement et en fonction de la réponse MRD au C1) avec suivi minimal de 3 ans. Temps de référence de 45 jrs (fin du C1) utilisé pour l’analyse des sous-groupes définis par l’obtention d’une RMC.
Résultats. 116 pts MRD+ traités. SG évaluée chez 110/116 pts atteints LAL-B Ph- et < 5 % blastes à l’inclusion, dont 74 ont reçu une greffe de CSH en RCh persistante. Suivi médian : 53,1 mois. SG médiane : 36,5 mois (IC95% : 22,0-NE), atteignant un plateau. Au moment de l’analyse, 30/74 (40,5 %) pts greffés en RCh persistante et 12/36 (33,3 %) pts non greffés sont en vie en RCh persistante. Analyse de l’impact de l’obtention d’une RMC après C1 effectuée chez 107 pts (1 évaluation non centralisée, 2 analyses avec sensibilité insuffisante). Chez ces pts, SG médiane non atteinte (IC95% : 27,3-NE) chez pts ayant obtenu une RMC (n = 85) et de 12,5 mois (IC95% : 3,2-39,7) chez pts n’ayant pas obtenu de RMC (n = 22 ; p = 0,002). Dans sous-groupe des pts greffés en RCh persistante, SG médiane depuis greffe non atteinte (IC95% : 25,7-NE) chez pts ayant obtenu une RMC (n = 61) et de 16,1 mois (IC95% : 1,1-NE) chez pts n’ayant pas obtenu de RMC (n = 10). Dans sous-groupe des pts en RC1, SG médiane non atteinte (IC95% : 29,5-NE) chez lpts ayant obtenu une RMC (n = 60) et de 10,6 mois (IC95% : 2,7-39,7) chez pts n’ayant pas obtenu de RMC (n = 13).
Conclusion. Chez pts adultes en RCh MRD+ traités par blin, la SG médiane atteint 36,5 mois avec un suivi médian à long terme de 53,1 mois, avec obtention d’un plateau. La SG médiane n’est pas atteinte chez les pts ayant obtenu une RMC après le C1 y compris chez les chez pts traités en RC1, de même que chez les pts ayant obtenu une RMC et ayant reçu une greffe de CSH en RCh persistante. Ces résultats confirment les bénéfices de survie à long terme associés au traitement de la MRD par le blinatumomab chez les adultes atteints de LAL-B.
06-07
Taux élevé de rémissions moléculaires obtenu par le blinatumomab chez des enfants ayant une leucémie aiguë lymphoblastique de la lignée B en rechute/réfractaire : RIALTO, programme d’accès rapide, multicentrique, en ouvert
B. Brethon*1, L. Franco2, G. Zugmaier3, P. Bader4, S. Jeha5, JP. Bourquin6, PG. Schlegel7, R. Handgretinger8, C. Rossig9, C. Chen-Santel10
1 Hématologie et Immunologie Pédiatrique, hôpital Robert Debré, AP-HP, Paris ; 2 Department of hematology/oncology and cell and gene therapy, IRCCS Bambino Gesù Children's Hospital, Rome, Italie ; 3 Département médical, Amgen Research (Munich) GmbH, München, Allemagne ; 4 Children and adolescents, University children's hospital Francfurt, Francfort, Allemagne ; 5 Oncology, St Jude Children's Research Hospital, Memphis, États-Unis ; 6 Pediatric oncology, Children's Research Center, Zurich, Suisse ; 7 Pediatrics, University Children's Hospital, Wuerzburg, Allemagne ; 8 Oncology, University Children's Hospital, Tuebingen, Allemagne ; 9 Oncology, University Children's Hospital, Muenster, Allemagne ; 10 Oncology, Charite University Médecine Berlin, Berlin, Allemagne
Introduction. Même si taux (tx) de guérison LAL pédiatriques élevé en première ligne, pronostic des LAL-B R/R reste sombre. Enfants avec LAL-B R/R ont reçu plusieurs traitements (ttt), les exposant à des toxicités aiguës avec séquelles à long terme limitant leur survie. Nouveaux ttts nécessaires pour contrôler la maladie et prolonger la survie avec une toxicité limitée. BLIN (BiTE®), anticorps bispécifique, redirige lymphocytes T CD3+ cytotoxiques vers cellules B CD19+. RIALTO évalue tolérance et efficacité du BLIN chez enfants avec LAL-B R/R.
Patients et méthodes. Patients pts) éligibles avec AL-B CD19+ R/R (≥ 2 rechutes, rechute post allogreffe cellules souches hématopoïétiques ou réfractaire à ttt antérieur) âgés entre 28 jours et 18 ans ; tx blastes médullaires ≥ 5 % ou < 5 % avec maladie résiduelle (MRD) ≥ 10-3. Ttt antérieur par BLIN autorisé si absence de maladie réfractaire à BLIN ou arrêt pour intolérance (réinclusion dans étude non autorisée). BLIN : 4 semaines perfusion continue/2 semaines sans, 5 cycles maximum : 15 μg/m2/j si tx blastes ≤25 % ; 5 μg/m2/j J1-J7 C1 puis 15 μg/m2/j si tx > 25 %. Suite ttt, dont allogreffe, non imposée par protocole et laissée à l’appréciation de l’investigateur. Critère principal : incidence des événements indésirables apparus pendant ttt (EIAT) et liés au ttt (EILT). Critères secondaires : réponse cytologique complète (RC ; < 5 % blastes), réponse moléculaire complète (MRD < 10-4 évaluée par PCR ou cytométrie de flux) lors C1 C2, survie sans rechute (SSR), survie globale (SG), tx allogreffes post BLIN.
Résultats. 98 pts d’âge médian 8,5 ans (4–17) inclus, 93 % en Europe ; 54 % avec tx blastes > 25 %, 41 % tx ≥ 50 % ; 44 % ont eu une allogreffe, 15 % une radiothérapie et 4 % BLIN ; 56 % des pts en ≥ deuxième rechute, 41 % rechuté post-allogreffe, 14 % primoréfractaires, 20 % réfractaires à réinduction pour première rechute. Au 09/03/2018, 37 pts encore dans RIALTO. 2 cycles reçus en médiane (1-5) et 4 pts ont reçu 5 cycles. EIAT chez 99 % des pts (6% grade (gr) ≥ 3). EILT chez 77 % des pts (26 % gr ≥ 3, 21 % graves). EIAT tous gr les plus fréquents : fièvre (83 %), vomissements (27 %), céphalées (24 %), anémie (19 %). EIAT d’intérêt tous gr/gr ≥ 3 (%) : réactions liées à la perfusion (67/9), infections (44/16), événements neurologiques (43/5), cytopénies (40/31), élévation des enzymes hépatiques (18/12), syndrome de relargage des cytokines (16/2), diminution des immunoglobulines (8/0), syndrome de lyse tumorale (4/2), syndrome de fuite capillaire (1/0). Interruption BLIN due à EILT chez 19 % des pts et arrêt définitif chez 4 %. 9 EI fatals, aucun lié au BLIN. Lors des C1 C2 : réponse globale chez 60 % des pts dont 40 % RC et 48 % réponse MRD. RC et réponse MRD chez les 2 pts avec t(17;19). 4 pts avec syndrome de Down (trisomie 21) : 3 réponses (2 RC et 1 réponse MRD). 4 pts déjà traités par BLIN : 3 RC dont 2 réponses MRD. 59 pts ont répondu après le C2, 27 (46 %) ont reçu une allogreffe, 19 ont rechuté et 5 sont décédés après suivi médian de 5,3 mois (0,3-13,2). SSR globale : 8,5 mois (IC95%, 2,9-NE) après réponse au BLIN avec suivi médian de 12,2 mois (0,5-14,1); 38 décès (32 liés à la LAL), SG médiane : 13,0 mois (IC95%, 9,3-NE).
Conclusion. Profil de tolérance du BLIN en monothérapie similaire à ceux d’autres essais chez enfants et adultes avec LAL-B R/R. BLIN efficace chez enfants avec LAL-B R/R : obtention réponse moléculaire complète chez près de 50 % des pts, dont les 2 pts avec une t(17;19) et la moitié de ceux déjà traités par BLIN. Meilleure réponse au BLIN si tx de blastes < 50 % avant BLIN. BLIN : option thérapeutique intéressante chez les enfants avec une LAL-B R/R.
06-08
Impact de l’hyperleucocytose sur la mortalité et la survie globale des patients adultes présentant une leucémie aiguë myéloblastique nouvellement diagnostiquée
C. Aboura*1, N. Boudjerra1, NEH. Guellouma1, Z. Kaci1, Y. Berkouk2, L. Metidji3, M. Belhani1
1 Service d’hématologie, CHU Beni Messous, Alger, Algérie ; 2 Service hématologie, CHU de Beni Messous, Alger, Algérie ; 3 Service d’hématologiebde, CHU Beni Messous, Alger, Algérie
Introduction. Dans la leucémie aiguë myéloblastique (LAM), 5 à 13 % des patients présentent une forme hyperleucocytaire, elle est définie par un taux de GB supérieur à 50 000/mm3.
Cette hyperleucocytose peut être à l’origine de graves complications telles que la leucostase, le syndrome de lyse tumorale (SLT) et la coagulation intravasculaire disséminée (CIVD) qui mettent en jeu le pronostic vital des patients et nécessitent une prise en charge immédiate et urgente.
Objectif. Evaluer l’impact de l’hyperleucocytose sur le pronostic des LAM.
Patients et méthodes. Il s’agit d’une étude rétrospective allant du 1er janvier 2014 au 31 décembre 2017, 102 patients (pts) atteints de LAM ont été colligés dont 24 ont présenté une forme hyperleucocytaire. Le recueil des données a été fait à partir des dossiers de malades hospitalisés. Les paramètres étudiés : âge, sexe, type cytologique, taux de GB au diagnostic, les complications liées à l’hyperleucocytose, le traitement symptomatique et le traitement de fond, la réponse au traitement, le devenir, les causes de décès et la survie globale.
Résultats. Sur une période de 4 ans, 102 diagnostics de LAM ont été posés dont 24 (23,5 %) ont présenté une forme hyperleucocytaire, parmi eux 7 pts (29 %) avaient une hyperleucocytose franche (taux de GB supérieur à 100 000/mm3). L’âge médian au diagnostic est de 42 ans (17-68). Sex-ratio H/F = 3,8. Après étude cytologique et phénotypique, le diagnostic de LAM4 ou LAM5 a été porté chez 15 pts (62,5 %). Les complications fréquemment retrouvées sont : SLT 13 pts (54 %). La CIVD 12 pts (50 %), La leucostase : 6 Pts (25 %) (Cérébrale : 1 pt, pulmonaire 5 pts). Le traitement a consisté chez tous les patients en une hyperhydratation avec Allopurinol. L’Hydréa a été prescrite dès le diagnostic à la dose de15 à 30 mg/kg/24 h. 4 pts (16,6 %) sont décédés avant traitement (3 pts par leucostase pulmonaire et 1 pt par leucostase cérébrale), tous ces patients avaient un taux de GB sup à 100 000. 20 pts (83,3 %) ont reçu une chimiothérapie d’induction.
Évaluation à la fin de l’induction : RC chez 2 pts (10 %), RP chez 12 pts (60 %), 9 pts décédés (4 par syndrome hémorragique, 3 par syndrome infectieux et 2 par SLT).
Au terme de notre étude et après un suivi médian de 48 mois, 7 patients (29 %) sont en RC dont 5 (21 %) ont été greffés, 2 perdus de vue avant le traitement d’induction (8 %), 2 pts (8 %) sont en rechute (après 15 mois et à 18 mois de RC).
La médiane de survie globale est de 9 mois. La survie globale à 15 mois est de 25 %, la survie sans progression à 15 mois est de 17 %.
Conclusion. L’hyperleucocytose dans la LAM constitue une urgence hématologique qui nécessite une prise en charge immédiate, il s’agit d’un facteur de risque important de morbidité et de mortalité précoce chez l’adulte atteint de LAM, la leucostase en est la principale complication.
06-09
Les leucémies aiguës myéloïdes du sujet âgé : caractéristiques clinicobiologiques, cytologiques, immunophénotypiques et cytogénétiques
H. MELLASSI*1, M. Châari1, M. Mdhaffar2, F. Imen3, I. Dammak1, F. Abida1, M. Elloumi2, H. Elleuch4
1 Laboratoire d’hématologie, CHU Hédi Chaker  , Sfax, Tunisie ; 2 Service d’hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 3 Hématologie clinique, chu Hédi Chaker, Sfax, Tunisie ; 4 Laboratoire hématologie, CHU Hédi Chaker, Sfax, Tunisie
, Sfax, Tunisie ; 2 Service d’hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 3 Hématologie clinique, chu Hédi Chaker, Sfax, Tunisie ; 4 Laboratoire hématologie, CHU Hédi Chaker, Sfax, Tunisie
Introduction. Le diagnostic des leucémies aiguës myéloïdes (LAM), une maladie dont l’incidence augmente avec l’âge, est basé essentiellement sur les données cytologiques des frottis sanguins et médullaires ainsi que les résultats de l’immunophénotypage (IPT). Nous nous sommes proposés de décrire les caractéristiques clinicobiologiques, cytologiques, immunophénotypiques et cytogénétiques d’une série de LAM de sujets âgés.
Patients et méthodes. Il s’agit d’une étude rétrospective réalisée au laboratoire d’hématologie de notre CHU s’étalant sur une période de 23 mois (de janvier 2017 à novembre 2018). Nous avons inclus les nouveaux cas de LAM de sujets âgés (> ou = de 55 ans), dont le diagnostic a été établi par étude cytologique du frottis sanguin et/ou du myélogramme, et par immunophénotypage (fait par un cytomètre BD Facs Canto II). Les aspects cytogénétiques ont été également notés.
Résultats. Nous avons colligé au total 30 cas de LAM. Une légère prédominance masculine a été notée (sex-ratio H/F = 1,14). La moyenne d’âge était de 63,8 ans avec des extrêmes allant de 55 ans à 85 ans. Un syndrome tumoral a été noté dans 33,3 % des cas. Une CIVD a été observée chez 3 patients. Une Thrombopénie a été trouvée dans 86,6 % et une anémie dans 83,3 %. La médiane des globules blancs était de 13 400 éléments/mm3 avec des extrêmes allant de 710 à 172 260 éléments/mm3. Une blastose sanguine a été notée chez 96,6 % des patients. Selon la classification FAB, nous avons observé 7 cas de LAM1, 5 cas de LAM2, 3 cas de LAM3, 5 cas de LAM4 et 10 cas (30 %) de LAM5. Selon la classification de l’OMS 2016, 8 cas (26,6 %) correspondaient à des LAM associées à des signes de myélodysplasie. L’IPT a permis de confirmer la nature myéloïde (expression de marqueurs myéloïdes communs CD13, CD33, MPOc) et elle était concordante avec la cytologie dans tous les cas. Chez 9 patients, une expression aberrante de marqueurs lymphoïdes était notée ; dont les plus fréquents : le CD7 (4 cas) et le CD56 (4 cas). Le caryotype a été réalisé chez 11 patients revenant pathologique dans 8 cas. Les anomalies notées étant : une translocation t(15;17) chez 2 cas de LAM3, une inversion (16) chez un cas de LAM4, et chez 5 cas de LAM associées à des signes de myélodysplasie ont a trouvé une del(5q) (un cas), une monosomie 7 (3 cas) et une trisomie 8 (un cas).
Conclusion. Notre travail montre que les LAM du sujet âgé sont fréquemment associées à des signes de myélodysplasie. En outre, elles sont douées d’un grand polymorphisme morphologique, immunophénotypique et cytogénétique d’où l’importance d’associer ces explorations dans la prise en charge de la maladie.
06-10
Évaluation prospective du FLT3 ligand plasmatique pendant l’induction de leucémie aiguë myeloide
P. Peterlin*1, J. Gaschet2, T. Guillaume1, A. Garnier1, M. Eveillard3, A. Le Bourgeois1, M. Cherel2, Y. Le Bris3, O. Theisen3, C. Touzeau1, T. Gastinne1, S. Le Gouill1, P. Moreau1, MC. Béné3, P. Chevallier1
1 Service d’hématologie clinique, CHU Hôtel-Dieu, Nantes ; 2 Université d’angers, université de Nantes, Nantes, France., CRCINA, INSERM, CNRS, Nantes ; 3 Service d’hématologie biologique, CHU Hôtel-Dieu, Nantes
Introduction. La cytokine Fms-like tyrosine kinase 3 Ligand (FL) joue un rôle clé dans l’hématopoïèse. Dans une précédente étude de phase 1 testant la radio-immunothérapie pour les leucémies aiguës lymphoblastiques en rechute/réfractaire (Lancet Haematol, 2015), il a été mis en évidence que seuls les patients répondeurs présentaient une augmentation de la concentration plasmatique du FL soluble (sFLc). Peu de données sont disponibles concernant l’évolution du sFLc durant le traitement des leucémies aiguës myéloïdes (LAM). Nous avons donc conduit une étude prospective monocentrique visant à évaluer l’impact de l’évolution du sFLc durant l’induction de LAM sur le devenir des patients. Nous avons pour cela inclus tous les patients atteint de LAM (hors LAM3) traités en première ligne et intensivement dans notre service durant une période de 18 mois.
Patients et méthodes. Les échantillons plasmatiques ont été prélevés aux jours 1/8/15/22 de l’induction, congelés puis analysés par ELISA (DY308, R&D Systems, Minneapolis, MN). L’impact du sFLc a été évalué sur le caractère réfractaire à l’induction, sur la rechute, sur l’EFS et sur l’OS.
Résultats. Entre mai 2016 et janvier 2018, 63 patients ont été inclus, 62 étaient analysables et 242 échantillons ont été analysés. Trois profils d’évolution du sFLc ont été mis en évidence : 1) augmentation constante du sFLc du J1 au J22 (groupe FLI (Increase)), 2) augmentation du J1 au J15 puis décroissance au J22 (groupe FLD (Deacrease)) et 3) stagnation à un bas niveau du sFLc (groupe FLL (Low)). L’âge médian était de 59 ans (29-71, < 60 ans n = 33). Le temps de suivi médian pour les patients en vie était de 541 jours (154-787). Vingt-six patients ont été classés dans le groupe FLI (42 %), 22 dans le groupe FLD (35 %) et 14 dans le groupe FLL (23 %). Les médianes de sFLc aux J1/8/15/22 étaient : FLI : 2, 724, 3673, 5 753 pg/mL ; FLD : 6, 1229, 6019, 684 pg/mL ; et FLL : 0, 60, 124, 81 pg/mL. Il n’y avait pas de différence entre les 3 groupes en ce qui concerne la classification ELN 2010, la classification OMS 2016, le taux de leucocytes ou le % de blastes médullaires au diagnostic. En analyse univariée, l’EFS et l’OS à 2 ans étaient significativement meilleures pour le groupe FLI (79,1±8 vs FLD 54,9 %±11 vs FLL 11,4 %±10, p < 0,0001 ; et 80,4 %±8 vs FLD 58,6 %±11 vs FLL 18,6 %±10, p = 0,09, respectivement). Il y avait une tendance à une association entre l’EFS à 2 ans et l’ELN mais pas pour l’OS (favorable : 70,9 %+-11, vs Int-1+Int-2 : 57,1 %+10 vs adverse 33 %+13, p = 0,06). La stratification des patients en fonction du sFLc médian à J15 (2952 pg/mL) et à J22 (1390 pg/mL) était associée à une EFS à 2 ans significativement différente : 38,2 %±9 pour les valeurs inférieures vs 71,8 %±8 pour les valeurs supérieures (p = 0,02) et 38,9 %+-9 vs 73,6 %+-8, (p = 0,02), respectivement. L’âge n’avait pas d’impact sur l’EFS ni sur l’OS. En analyse multivariée, en considérant l’âge, l’ELN, le sFLc aux J15 et J22 et le profil de cinétique d’évolution du sFLc (FLI vs FLD vs FLL), ce dernier était le facteur le plus fortement associé à l’EFS (HR : 3,62 ; 95 %CI : 1,65-7,94, p = 0,001; ELN : HR : 1,74 ; 95 %CI : 0,98-3,10, p = 0,05; sFLc àJ15 p = 0,37; sFLc àJ22, p = 0,24, âge p = NS) ; et le seul à être significativement associé à l’OS (HR : 2,60 ; 95 %CI : 1,12-6,07, p = 0,02).
Conclusion. Le profil de cinétique d’évolution du sFLc durant l’induction apparaît comme un nouveau puissant outil pronostic pour les patients traités pour une LAM. Cela nécessite une validation sur une plus grande cohorte et les mécanismes sous-jacents restent à être élucidés.
06-11
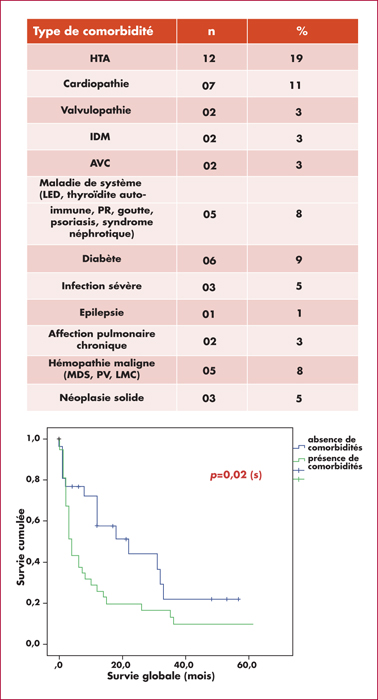
Impact des comorbidités sur le pronostic des patients présentant une leucémie aiguë myéloblastique
F. Talbi*, R. Abbadi, A. Ghassoul, M. Djilali, K. Belateche, L. Bouteldja, S. Belakehal, FZ. Ardjoun, K. Djouadi
hématologie, hôpital Central De l’Armée DR Mohammed Seghir Nekkache, Alger, Algérie
Introduction. Les leucémies aiguës myéloblastiques (LAM) ont un pronostic péjoratif, en particulier chez le sujet âgé, en raison de la présence de comorbidités rendant la prise en charge de ces patients particulièrement délicate. La décision de mettre en route une chimiothérapie intensive (seul espoir pour améliorer la survie des patients) doit se discuter en comité multidisciplinaire vu que la mortalité liée au traitement est très importante dans cette population de malades.
Patients et méthodes. Nous avons inclus rétrospectivement les patients présentant une LAM diagnostiquée au service d’Hématologie de l’HCA sur une période de 5 ans (2013-2018). Nos patients ont bénéficié d’un interrogatoire, un examen clinique, un hémogramme systématique, un frottis sanguin et médullaire, une cytométrie en flux ; la cytogénétique étant d’introduction récente, cette variable n’a pas été évaluée dans notre étude). Nous avons calculé le risque de décès ou RR à partir de notre cohorte (exposé/non exposé). La survie globale à 5 ans est estimée par la méthode de Kaplan Meier et la comparaison des courbes par le test du Logrank.
Résultats. n = 64 (évaluables pour le paramètre étudié), n = 40 H/ 24 F, sex-ratio = 1,66 ; l’âge moyen = 46 ans ± 19 [14-82]. n = 27 patients présentent au diagnostic des comorbidités : les principales tares observées dans notre série sont : résumées dans le tableau suivant. La survie médiane est de 24 mois [IC95% 15,61-33,67] dans le groupe de patients sans antécédent vs 13 mois [IC95% 6,61-18,8]dans le groupe avec tare associée : p = 0,02 (figure 1).

Conclusion. Les comordités assombrissent considérablement le pronostic des LAM comme le montrent les résultats de notre étude, ce qui rejoint les données de la littérature.
06-12
Les facteurs pronostiques des leucémies aiguës myéloblastiques
F. Talbi*, L. Bouteldja, R. Abbadi, M. Djilali, A. Ghassoul, K. Belateche, S. Belakehal, FZ. Ardjoun, K. Djouadi
hématologie, hôpital Central De l’Armée DR Mohammed Seghir Nekkache, Alger, Algérie
Introduction. Nous nous sommes proposés d’évaluer l’impact pronostique de huit facteurs retenus par de nombreuses études comme étant étroitement liés au devenir des patients atteints d’une LAM.
Matériels et méthodes. Nous avons évalué rétrospectivement l’impact : de l’âge > 65 ans, du sexe, du délai diagnostique, de la présence d’une infection (au diagnostic), de l’hyperleucocytose > 50 000 elts/mm3, de la blastose périphérique, du taux de LDH (> 400 UI/L) et du traitement d’induction instauré (chimiothérapie vs traitement palliatif) sur l’évolution des patients.
Notre cohorte inclut tous les patients présentant une LAM diagnostiquée au service d’Hématologie de l’HCA sur une période de 17 ans (2000-2018). Les patients ont bénéficié d’une exploration hématologique clinique et biologique (HG, FS, MO, CCC au noir soudan ± à la myéloperoxydase, une CMF ainsi qu’un bilan biochimique complet). Nous avons calculé les RR de ces différentes variables en appliquant la régression logistique.
Résultats. N = 180 (évaluables), n = 120 hommes/60 femmes, sex-ratio = 2 ; l’âge moyen = 50 ans ± 20 [14-91], il est > 65 ans dans n = 49 cas (27 %).
Le délai moyen du diagnostic = 32 ± 30 jours [1-240], il est > 30 jours dans 75 cas (42 %).
Une infection est retrouvée (au diagnostic) dans 83 cas (46 %).
Le taux moyen de GB = 45.255 ± 6 868 elts/mm3 [300-707 970], il est > 50 000 elts/mm3 dans 49 cas (27 %).
Le taux de blastes périphériques est en moyenne de 46 ± 32 % [0-100].
Le taux moyen de LDH est = 747 ± 320 UI/L [107-7,343], il est > 400 UI/L dans n = 61 cas (34 %).
N = 122 patients ont bénéficié d’une chimiothérapie d’induction (68 %), n = 91 patients (50 %) ont reçu un protocole avec trois injections d’anthracyclines associées à la cytarabine (protocole type 3+5, 3+7 ou 3+10).
Le risque de décès étudié en fonction de ces différentes variables retrouve : p = 0,23 (NS) pour l’âge > 65 ans (n = 102) vs ≤ 65 ans (n = 42 décès) ; p = 0,59 (NS) pour le sexe (n = 47 décès/sexe féminin vs n = 97/sexe masculin) ; p = 0,55 (NS) pour le délai diagnostique (n = 67 décès si délai < 30 j contre n = 57 si délai ≥ 30 jours) ; p = 0,88 (NS) pour le risque de décès chez les patients présentant une infection « baseline » : n = 78 décès vs n = 65 en présence de ce facteur ; p = 0,53 (NS) en fonction de la présence ou non d’une hyperleucocytose > 50 000 elts/mm3 (n = 39 vs 104) ; p = 0,02 (S) LDH > versus≤ 400 UI/L ; p = 0,33 (NS) pour le risque de décès en fonction du pourcentage de blastes périphériques ; p = 0,25 (NS) pour la CT d’induction (n = 69 décès dans chaque groupe.
Les chances d’obtenir une RC en fonction du délai diagnostique :p = 0,03 (S), du sexe p = 0,03 (S), de l’âge p = 0,000 (S), du taux de LDH > 400 UI/L (p = 0,048) et du type de CT d’induction p = 0,000 (S)/.
Conclusion. Le risque de décès est étroitement lié au taux de LDH dans notre étude, les autres facteurs sont non significatifs sur cette évaluation ; en revanche, le pourcentage de RC est corrélé positivement au délai diagnostique, au sexe féminin, à l’âge < 65 ans, à la CT d’induction (en plus du taux de LDH).
06-13
Érythroleucémie à propos de 10 cas
F. Talbi*1, R. Abbadi1, K. Belateche1, L. Bouteldja1, M. Djilali1, A. Ghassoul1, M. Boudehar2, S. Menouer1, S. Belakehal1, FZ. Ardjoun1, K. Djouadi1
1 hématologie, hôpital Central De l’Armée DR Mohammed Seghir Nekkache, Alger, Algérie ; 2 Réanimation néonatale, hôpital Central De l’Armée DR Mohammed Seghir Nekkache, Alger, Algérie
Introduction. La leucémie aiguë érythroblastique (LAM-M6) est une entité rare, représente 3 à 4 % de l’ensemble des LA. Il en existe deux types : l’érythroleucémie et la leucémie érythroïde pure. Elle se manifeste le plus souvent par des signes de cytopénie et d’infiltration des tissus extra-hématopoïétiques, elle est plus fréquente chez les adultes que chez les enfants avec un pronostic péjoratif.
Patients et méthodes. Le diagnostic est retenu sur un examen cytologique du frottis sanguin (FS) et médullaire (PMO) ainsi que l’examen immunophénotypique. Nous avons calculé la survie globale par la méthode de Kaplan Meier.
Résultats. N = 181 (évaluables), n = 10 patients présentant une LAM6 selon la classification FAB (5 %). • L’étude cytologique du FS et PMO+ résultats de la CMF ont conduit au diagnostic de l’érythroleucémie chez 8 de nos patients et de leucémie érythroïde pure chez 2 patients. L’âge moyen = 47 ± 21,77 ans [0-64]. Le sex-ratio = 1,5 (6 H/4 F). Le délai dc : 40 ± 35 j [01-90]. Des signes d’insuffisance médullaire : PCM (100 %), syndrome hémorragique : 3 cas (30 %), une infection : 4 cas (40 %), une splénomégalie : 4 cas (30 %). Le taux moyen de GB = 84,300 ± 11,260 G/L [125,000-83,400], le taux de blastes périphériques (moy = 26 ± 23%[1-65]), blastes médullaires = moy 49,5 ± 19 % (8-93]. HBmoy = 8, 06 ± 1, 9 g/dL [5-10], PLQ : 21-151 G/L (moy : 65,900 ± 47,450 elts/mm3). N = 3 patients ont présenté une thrombopénie sévère à 13-21-39 G/L respectivement (30 %). Une pancytopénie retrouvée dans n = 5 cas (50 %). LDH (moy) = 905 ± 896 UI/L [12-2,900], n = 2 patients ont présenté 1 complication métabolique au diagnostic (hyperkaliémie, hyponatrémie). N = 8 patients ont bénéficié d’une CT d’induction (3+7), n = 1 patient n’a reçu que 2 injections d’anthracyclines associées à la cytarabine. Nous avons diagnostiqué une LAM- NOS selon la classification OMS 2008. Nouveau né (H18) : clinique : PCM+hépatosplénomégalie, biologie : GB : 125 G/L, (EB = 64%, blastes = 3%), HB = 10 g/dL, PLQ = 13 G/L, PMO : infiltration par 68 % d’EB dystrophiques++++, 8 % de MB, le NNe est sous aspirateur artificiel. Le TRT spécifique n’a pas encore été débuté La RC obtenue (n = 4 cas (40 %)) et n = 3 échecs (30 %), n = 1 patient sorti contre avis médical ; un décès en induction répertorié (toxicité cardiaque aiguë). La SG = 27% (figure 1).
Conclusion. Nos données rejoignent celles de la littérature : la LAM6 pathologique rare (3-5/séries, 5 % dans la nôtre), exceptionnelle chez l’enfant. Les signes d’insuffisance médullaire sont quasi constants, 1 pancytopénie dans 50 %. Une splénomégalie est retrouvée dans un 1/3 des cas. Le pronostic est sombre (SG = 25% à 5 ans), d’où l’intérêt d’un diagnostic rapide et d’une prise en charge adéquate.
06-14
Les leucémies aiguës myéloïdes 3 au niveau de l’Ouest algérien : étude multicentrique
N. Mehalhal*1, A. Krim2, F. Serradj2, H. Gaid3, A. Bekadja4, B. Benzineb5, N. Mesli6, S. Zouani7, D. Saidi7, L. Zatla8, H. H. Touhami7, M. Aberkane9, A. Bachiri10, M. Benlazar11, Z. Zouaoui12
1 Hématologie, EPH Mascara, Mascara, Algérie ; 2 Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie ; 3 Hématologie, Établissement Hospitalier Universitaire d’Oran, Oran, Algérie ; 4 Hématologie et Thérapie Cellulaire, Établissement Hospitalier Universitaire 1er-Novembre, Oran, Algérie ; 5 Hématologie, CHU Tlemcen Faculté de médecine Tlemcen, Tlemcen, Algérie ; 6 Hématologie, CHU Dr Tidjani Damardji, Tlemcen, Algérie ; 7 Hématologie, CH et Universitaire d’Oran, Oran, Algérie ; 8 Hématologie, CHU Oran, Oran, Algérie ; 9 Hématologie, Hôpital Militaire Régional Universitaire d’Oran, Oran, Algérie ; 10 Hématologie, HMRU Oran, Oran, Algérie ; 11 Hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Algérie ; 12 Service d’hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Algérie
Introduction. La LAM3 est une hémopathie maligne relativement rare, caractérisée par une t(15;17) responsable de l’activation du gène RARE alpha. Son pronostic a été complètement bouleversé par l’apport de l’Acide tout-transrétinoïque et l’arsenic, qui ont amélioré autant les taux de réponse que les survies. L’objectif de ce travail est d’évaluer la prise en charge des LAM3 au niveau de l’Ouest Algérien.
Patients et méthodes. Étude rétrospective multicentrique, de 2007 à 2017. Six services d’Hématologie y ont participé. Ont été inclus tous les pts adultes âgés > 15 ans et atteints d’une LAM3. Le diagnostic a été établi à l’aide du myélogramme, la cytochimie, la CMF et dans quelques cas par la cytogénétique à la recherche de la t(15;17). Le traitement de type « 3+7 » associé ou pas à l’acide tout-transrétinoïque (ATRA) à la dose de 45 mg/m2/j jusqu’à l’obtention de la RC. L’entretien à base d’ATRA à la même dose pendant une durée de 2 années. L’évaluation a porté sur les taux de RC et sur les survies sans rechute et survies globales.
Résultats. Sur une période de 11 ans (2007 à 2017), 34 pts ont été colligés, d’âge médian = 38 ans (15-73), dont H21/F13 avec un sex-ratio = 1, 6. Le PS (0-1) = 14 (41%); (>2)=16(18%); (>3)=4(12%). Le tableau clinique est marqué par un syndrome tumoral chez 25 (73 %) pts, un syndrome anémique chez 27 (79 %) pts, un syndrome infectieux chez 11 (32 %) pts et un syndrome hémorragique chez 22 (65 %) pts. Sur le plan biologique, le taux de GB < 4 G/L = 12 (%); [4-10 G/L] = 4 (%); >10 G/L = 16 (%), le taux de GB médian = 9,5 G/L (0,7-800), le taux d’Hb médian = 7,5 g/dL (4-11,7), le taux de plaquettes médian = 27,5 G/L (2-119). Le taux médian de la blastose sanguine = 78% (15-100), celui des blastes médullaires = 80% (33-100). Le syndrome de CIVD a été retrouvé chez 32 % des pts. Selon la classification FAB, le type M3 = 30 (88%) et la M3 variant = 4 (12%). La t(15;17) a été recherchée chez seulement 3/34 (9 %) des pts. Selon le score pronostic de SANZ, 50 % des pt sont de score pronostic élevé. Du point de vue thérapeutique, TRT « 3+7 »+ATRA = 26/34=76%, TRT « 3+7 » seul = 4/34=12% et pts non traités = 4/34=12%. Parmi les pts traités par « 3+7 » + ATRA (26 pts), les résultats montrent : décès précoces = 03/26=12% (non évaluables pour la réponse), RC = 19/23= 83%, Échec = 4/23=17%, rechute = 10/19=53%. En termes de survies, avec un suivi moyen de 25 mois, la médiane de SG = 70 mois ; 29 % à 72 mois et la médiane de SSR = 34 mois ; 32 % à 72 mois. La SG selon le score pronostic de Sanz, montre une différence significative entre les risques faible, intermédiaire et élevé.
Conclusion. La fréquence de laLAM3 est de 5 % dans notre étude sur un total de 666 DE LAM. Cette étude est un état des lieux de la prise en charge des LAM au niveau de l’Ouest Algérien. Les résultats montrent une insuffisance des moyens du diagnostic (cytogénétique et biologie moléculaire) et des moyens de la réanimation hématologique, vu le taux élevé des décès précoces, en particulier liés à la CIVD et aux hémorragies. 50 % présentent au diagnostic, un score pronostic de SANZ de risque élevé, témoignant de la gravité de la maladie au diagnostic. 21 % des pts, n’ont pas bénéficié de l’ATRA, ce qui pose le problème de l’équité à l’accès aux soins pour tous les patients. L’apport de l’ATRA a permis de transformer le pronostic de la LAM3 dans sa prise en charge locale, avec une amélioration nette des taux de réponse et de survie.
06-15
Étude du profil moléculaire des leucémies aiguës myéloïdes dans la population adolescents et jeunes adultes (15-25 ans)
L. Fenwarth*1, N. Boissel2, H. Lapillonne3, A. Marceau-Renaut1, E. Lengline2, C. Récher4, N. Duployez1, A. Baruchel5, G. Michel6, E. Jourdan7, X. Thomas8, H. Dombret9, G. Leverger10, A. Petit11, C. Preudhomme1
1 Laboratoire d’hématologie, CHU Lille, Lille ; 2 Département d’hématologie, Hôpital Saint-Louis, AP-HP, Paris ; 3 Laboratoire d’hématologie, Hôpital pour enfants Trousseau, Paris ; 4 Hématologie adulte, Institut Universitaire du Cancer de Toulouse-Oncopole, Toulouse ; 5 Hématologie et immunologie pédiatrique, hôpital Robert Debré, AP-HP, Paris ; 6 Service d’hématologie et oncologie pédiatrique, Hôpital de la Timone, Marseille ; 7 Hématologie, CHU, Nîmes ; 8 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 9 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 10 Service d’hématologie et oncologie pédiatrique, Hôpital d’enfants Armand Trousseau, AP-HP, Paris ; 11 Service d’oncologie et hématologie pédiatrique, Hôpital pour enfants Trousseau, Paris
Introduction. La population adolescents et jeunes adultes (AJA) se situe à la frontière entre la population pédiatrique et la population adulte, et s’est distinguée au cours des dernières années en oncohématologie en raison de ses caractéristiques propres. Le sous-type « Philadelphie-like » des leucémies aiguës lymphoblastiques est ainsi plus commun chez les AJA que dans la population pédiatrique. Les leucémies aiguës myéloïdes (LAM) constituent un groupe d’hémopathies malignes hétérogène sur le plan cytogénétique et moléculaire, et varie avec l’âge. Il s’agit d’une population dont le profil mutationnel demeure encore peu caractérisé. L’objectif de cette étude était de définir le profil moléculaire des LAM de la population AJA.
Patients et méthodes. Cette étude rétrospective multicentrique inclut l’ensemble des AJA, âgés de 15 à 25 ans, présentant une LAM de novo issus des protocoles ELAM02 (#NCT00149162), CBF-2006 (#NCT00428558) et ALFA-0702 (#NCT00932412). L’étude du profil mutationnel des AJA a été réalisée par technique de séquençage à haut débit sur des librairies HaloplexTM (Agilent®) et/ou AmpliSeqTM (Thermo Fisher®). Le profil mutationnel des AJA a été comparé à celui de la cohorte pédiatrique (0-14,9 ans) de l’ELAM02.
Résultats. Un total de 140 patients AJA a été inclus dans l’étude. Une mutation était retrouvée chez 84,3 % des patients, avec un nombre médian de mutations de 2 par patient (de 0 à 7 par patient). Les mutations des gènes régulant les voies de signalisation étaient les plus fréquents (NRAS : 27,1 %, FLT3-ITD : 20,7 %, FLT3-TKD : 11,4 %, KRAS : 8,6 %, KIT : 7,9 %). En comparaison à la population pédiatrique, les AJA avaient une charge mutationnelle plus importante (nombre médian de mutations chez l’enfant : 1 par patient, p = 0,014). Les mutations des gènes CEBPA (14,3 %) ASXL1 (7,9 %), DNMT3A (5,0 %), du complexe cohésine (10,7 %) ainsi que de type KMT2A-PTD (5,0 %) et FLT3-ITD (20,7 %) étaient significativement plus fréquentes chez les AJA. La fréquence des mutations FLT3-TKD (11,4 %), IDH1 (3,6 %) et IDH2 (4,3 %) n’était pas statistiquement différente entre les AJA et la population pédiatrique. La survie globale à 5 ans de la population AJA n’était pas statistiquement différente de celle de la population pédiatrique (73,6 % vs 73,5 %, p = 0,969) et n’était pas influencée par le type de traitement (adulte vs pédiatrique, p = 0,469). La classification ELN 2017 (1) permettait de stratifier la population AJA en 3 groupes de risque, mais les groupes intermédiaire et défavorable ne pouvaient être distingués (p = 0,639). L’utilisation d’un classifier moléculaire évalué dans la population pédiatrique par Marceau-Renaut et al. (2) permettait de discriminer les 3 groupes de risque de façon statistiquement significative au sein des AJA (p < 0,001).
Conclusion. Les AJA présentent un profil moléculaire s’inscrivant dans un continuum entre celui de la population pédiatrique et celui des adultes plus âgés. Les mutations sont plus fréquentes dans la population AJA, en particulier avec l’émergence de gènes de l’épigénétique ce qui offre la possibilité de mieux stratifier cette population. Enfin, étant donné la fréquence plus élevée des mutations de FLT3 chez les AJA, ceux-ci pourraient bénéficier des nouvelles thérapeutiques ciblées (inhibiteurs de FLT3).
06-16
Évaluation de l’incidence des syndromes de prédisposition génétique aux leucémies aiguës myéloïdes pédiatriques issues du protocole ELAM02
L. Fenwarth*1, N. Duployez1, A. Marceau-Renaut1, W. Abou Chahla2, S. Ducassou3, V. Gandemer4, M. Pasquet5, T. Leblanc6, P. Schneider7, C. Domenech8, P. Saultier9, G. Leverger10, H. Lapillonne11, C. Preudhomme1, A. Petit12
1 Laboratoire d’hématologie, CHU Lille, Lille ; 2 Service d’hématologie pédiatrique, CHU Lille, Lille ; 3 Service d’hématologie et oncologie pédiatrique, Hôpital des Enfants, Bordeaux ; 4 Hématologie pédiatrique, CHU de Rennes, Rennes ; 5 Service d’hématologie et immunologie pédiatrique, Hôpital pour enfants, Toulouse ; 6 Service d’hématologie pédiatrique, Hôpital Robert-Debré, Paris ; 7 Service d’hémato-oncologie pédiatrique, CHU Rouen, Rouen ; 8 Équipe « Migration et différenciation des cellules souches hématopoiétiques », Laboratoire de biologie du développement, CNRS UMR 7622, INSERM U 1156, IBPS, UPMC, Paris ; 9 Service d’hématologie oncologie pédiatrique, Hôpital de la Timone Enfant, Marseille ; 10 Service d’hématologie et oncologie pédiatrique, Hôpital d’enfants Armand Trousseau, AP-HP, Paris ; 11 Laboratoire d’hématologie, Hôpital pour enfants Trousseau, Paris ; 12 Service d’oncologie et hématologie pédiatrique, Hôpital pour enfants Trousseau, Paris
Introduction. Le séquençage de haut débit (next generation sequencing [NGS]) a permis de mieux apprécier le profil moléculaire des leucémies aiguës myéloïdes (LAM) au cours des dernières années. Les mutations identifiées ont contribué à une meilleure stratification pronostique et ont permis d’optimiser le traitement des patients. Néanmoins le caractère somatique ou germinal des mutations n’est pas recherché en routine en l’absence d’histoire familiale ou de signes d’appel cliniques évocateurs. L’identification des syndromes de prédisposition aux cancers de l’enfant constitue un enjeu majeur et a été intégré dans le premier axe du PAIR pédiatrie. Ainsi, l’objectif de cette étude était d’évaluer l’incidence des syndromes de prédisposition génétique aux LAM sans manifestation clinique extrahématologique associée au sein de la population pédiatrique incluse dans le protocole ELAM02.
Patients et méthodes. Il s’agit d’une étude rétrospective multicentrique incluant l’ensemble de la population pédiatrique présentant une LAM de novo issue du protocole ELAM02 (#NCT00149162) et analysée en NGS (1). L’ensemble des patients présentant une mutation biallélique ou retrouvée avec une fréquence allélique (variant allele frequency [VAF]) élevée (≥ 30 %) en séquençage de haut débit dans l’un des cinq gènes suivants : GATA2, CEBPA, RUNX1, TP53, ETV6 était inclus. Le caractère constitutionnel de la mutation était confirmé par la persistance de la mutation en séquençage Sanger sur le prélèvement sanguin périphérique ou médullaire des patients lors de la rémission complète (RC).
Résultats. Au sein des 385 patients issus du protocole ELAM02 analysés en NGS, un total de 44 patients a été inclus dans cette étude, et l’analyse du prélèvement en RC a pu être réalisée chez 33 d’entre eux. Sur les 5 gènes analysés, 17 patients présentaient une mutation biallélique de CEBPA (dont une mutation homozygote), et 12 patients une mutation de GATA2. Quatorze patients présentaient une mutation de RUNX1 parmi lesquels 3 avaient une mutation biallélique et 11 une mutation avec VAF supérieure à 30 %. La mutation d’ETV6 était retrouvée chez deux patients de même que celle de TP53. Au total 4 patients présentaient une mutation persistant en RC, incluant : une mutation de CEBPA, deux de GATA2 et une de RUNX1, suggérant le caractère constitutionnel de ces mutations.
Conclusion. La présence de mutations bialléliques ou d’une fréquence allélique élevée constitue de nouveaux points d’appel pour le diagnostic de syndrome de prédisposition aux LAM. L’étude des altérations moléculaires au diagnostic de LAM constitue ainsi un outil performant pour la détection des syndromes de prédisposition aux hémopathies myéloïdes sans manifestation extra-hématologique associée et devrait être intégrée dans la pratique clinique quotidienne. L’identification de syndromes de prédisposition aux LAM revêt d’un intérêt majeur dans la prise en charge des patients en particulier lorsque le suivi de la MRD sur une mutation est envisagé (2), nécessitant l’évaluation préalable du caractère somatique ou germinal de cette dernière. Le développement du profilage moléculaire des LAM par NGS est amené à identifier plus fréquemment des altérations germinales. La formalisation du circuit de rendu des résultats au patient doit faire l’objet d’une réflexion avec les médecins cliniciens.
06-17
Régulation et fonctions du micro-ARN16 dans les leucémies aiguës myéloïdes FLT3-ITD
G. Sueur*1, A. Boutet1, S. Manenti2, S. Bertoli3
1 Centre de recherches en cancérologie de Toulouse, INSERM, Toulouse ; 2 Centre de recherches en cancérologie de Toulouse, CNRS, Toulouse ; 3 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse
Introduction. Les patients présentant une Duplication Interne en Tandem dans le récepteur FLT3 (FLT3-ITD) représentent 25 % des cas de leucémie aiguë myéloïde (LAM), et ont un mauvais pronostic de survie par rapport aux patients exprimant le récepteur sauvage. En effet, les LAMs FLT3-ITD ont tendance à rechuter après les traitements conventionnels, ainsi qu’après des thérapies ciblant FLT3. Il est donc nécessaire d’étudier plus en détail la biologie de ce sous-type de LAM, afin de déterminer de meilleurs moyens de cibler ces cellules.
Notre équipe a précédemment identifié CDC25A comme un acteur indispensable dans la prolifération et le blocage de différenciation de ces cellules, deux caractéristiques principales des LAM. Cette même étude a mis en évidence un contrôle strict du niveau de CDC25A par le récepteur muté. CDC25A apparaît donc comme une cible potentiellement très intéressante dans le traitement de ces leucémies, mais la conformation de cette phosphatase la rend difficile à inhiber pharmacologiquement. Nous souhaitons donc nous intéresser plus en détail à la voie de signalisation FLT3-ITD/CDC25A afin de trouver une autre approche pour la cibler.
Résultats. Dans ce travail, nous montrons tout d’abord que dans les cellules de LAM FLT3-ITD STAT5 constitue un régulateur transcriptionnel de CDC25A. Nous avons ensuite décidé d’étudier le rôle du suppresseur de tumeur microARN-16 (miR16) dans la régulation de CDC25A par FLT3-ITD, dans la mesure où il a été montré dans d’autres modèles que miR16 était i) capable de diminuer le niveau d’ARNm de CDC25A et ii) invalidé par le récepteur FLT3-ITD. Par des expériences d’interférence à l’ARN et l’utilisation d’inhibiteurs pharmacologiques, nous avons montré une inhibition de miR16 par FLT3-ITD, via le facteur de transcription STAT5. De plus, une inhibition du miR16 suffit à rétablir l’expression de CDC25A après inhibition de FLT3 ou invalidation de STAT5, ce qui illustre l’importance de la répression du miR16 pour maintenir le niveau de CDC25A dans ces cellules leucémiques.
Nous nous sommes ensuite intéressés au rôle fonctionnel du miR16, que nous avons à cet effet surexprimé dans nos cellules leucémiques. Cette surexpression conduit à un arrêt de prolifération et une induction de la différenciation des cellules FLT3-ITD, ainsi que de cellules FLT3-ITD/TKD résistantes à l’inhibition de FLT3. Les cellules leucémiques FLT3 sauvage sont par contre insensibles à cette surexpression. Par ailleurs, des expériences d’inhibition du miR16 nous ont permis de montrer que l’augmentation de son expression constituait un facteur important de l’arrêt de prolifération causé par un inhibiteur de FLT3 dans les cellules FLT3-ITD.
Conclusion. En résumé, nous avons identifié le micro-ARN16 comme un régulateur négatif clé de CDC25A en aval de la voie FLT3-ITD/STAT5, et mis en évidence son rôle de suppresseur de tumeur dans les LAMs FLT3-ITD, où sa réexpression permet un arrêt de prolifération ainsi qu’une reprise de la différenciation dans les cellules leucémiques exprimant FLT3-ITD.
06-18
Apport de la RT-MLPA dans les leucémies aiguës myéloïdes de l’adulte : Analyse rétrospective du protocole ALFA-0702
B. Ducourneau*1, C. Terré2, A. Daudignon3, P. Ruminy4, JM. Cayuela5, I. Luquet6, S. Hayette7, D. Bories8, D. Naguib9, P. Etancelin10, R. Kim11, J. Vargaftig12, I. Vaida13, S. De Botton14, X. Thomas15, C. Preudhomme16, N. Duployez16
1 Laboratoire d’hématologie, Ch de Valenciennes et CHRU de Lille, Lille ; 2 Laboratoire de cytogénétique, Centre Hospitalier de Versailles, Le Chesnay ; 3 Laboratoire de cytogénétique Jeanne de Flandres, CHU Lille, Lille ; 4 Inserm U918, Centre Henri Becquerel, Rouen ; 5 Laboratoire d’hématologie, Hôpital Saint-Louis, Paris ; 6 Laboratoire d’hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 7 Biologie moléculaire, CHU, Lyon ; 8 Laboratoire de biologie moléculaire, Hôpital Henri Mondor, Créteil ; 9 Laboratoire d’Hématologie, CHU Côte de Nacre, Caen ; 10 Laboratoire de Génétique Oncologique, Centre Henri Becquerel, Rouen ; 11 Hématologie biologique, Hôpital Saint-Louis AP-HP, Paris ; 12 Division of hematology, Curie Institute, Saint-Cloud ; 13 Hématologie, CHR Pontoise, Pontoise ; 14 Service d’hématologie clinique, Institut Gustave Roussy, Villejuif ; 15 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 16 Laboratoire d’hématologie, Inserm UMRS 1192, CHU de Lille, Lille
Introduction. La cytogénétique dans les LAM est indispensable chez les enfants et chez les adultes. Elle permet de séparer en 3 groupes pronostiques les patients atteints de LAM : favorable, intermédiaire et défavorable. Celle-ci permet notamment de détecter les translocations récurrentes qui sont un mécanisme privilégié d’activation d’oncogènes dans les tumeurs hématopoïétiques et particulièrement fréquentes dans les LAM. Les techniques de cytogénétique reposent sur le caryotype mais également sur l’hybridation in situ fluorescente (FISH) qui peut être réalisé en cas d’échec de ce dernier et de recherche d’anomalies cryptiques. Toutefois ces transcrits chimériques peuvent également être détectés par RT-MLPA. Dans cette étude, nous avons évalué l’impact de la RT-MLPA sur une grande cohorte de patient : le protocole ALFA-0702.
Patients et méthodes. La RT-MLPA est réalisée grâce au design de 240 oligonucléotides localisés sur 68 gènes. Après préparation de l’ADNc, celui-ci est hybridé avec le mélange suivi d’une ligation et enfin d’une amplification par PCR et du séquençage par pyroséquenceur. Dans cette étude, nous avons testé 566/713 prélèvements disponibles au diagnostic du protocole ALFA-0702.
Résultats. Concernant les 566 patients analysés, la RT-MLPA a permis de confirmer les anomalies moléculaires détectées par cytogénétique et biologie moléculaire classique : 18 duplications de MLL, 6 DEK-CAN, 1 CALM-AF10, 18 réarrangements de MLL, 5 NPM1-MLF1, 1 MOZ-CBP. Toutefois, un réarrangement MLL-ENL n’a pas pu être détecté car les sondes du transcrit impliqué ne sont pas présentes dans le mix.
Par ailleurs, cette technique nous a permis d’identifier en plus 5 CBFβ-MYH11, 2 DEK-CAN, 1 MOZ-CBP, 1 duplication de MLL, 3 réarrangements de MLL et 9 NUP98-NSD1.
Sur le plan pronostique, l’impact majeur de cette étude est l’identification du transcrit NUP98-NSD1 dont l’incidence est ici de 1,6 % dans cette étude, avec un taux de rechute de 66 % et une survie médiane de 22 %.
Par ailleurs, une révision cytogénétique de nos résultats de RT-MLPA a permis d’expliquer 2 cas rares : un patient présentant une insertion de MLL dans AF10 difficilement détectable par FISH en sonde Break-apart MLL mais mise en évidence par RT-MLPA ; un patient présentant un clone majoritaire avec caryotype complexe (et une del(5q)) donc de pronostique défavorable et un clone minoritaire avec une insertion de CBFβ dans MYH11 donc de pronostique favorable difficilement détectable par FISH.
Conclusion. La détection par RT-MLPA des transcrits de fusion dans les LAM est un outil très intéressant à mettre en place en routine car c’est une technique de biologie moléculaire simple et rapide. Sa mise en place en routine au laboratoire de biologie moléculaire du CHRU de Lille permet de rendre en 48 h la recherche des transcrits de fusion dans les LAM, qu’il s’agisse de transcrit connus ou plus rares. Par ailleurs, grâce à notre étude sur le protocole ALFA-0702 nous avons pu montrer qu’elle permet de reclasser un certain nombre de patients. Il serait notamment intéressant de rechercher systématiquement le transcrit NUP98-NSD1 dans les LAM de l’adulte qui semble être associé à un pronostic très défavorable.
06-19
Traitement de rattrapage par fludarabine, cytarabine et gemtuzumab ozogamicine dans les leucémies aiguës myéloblastiques de l’enfant réfractaires à la première ligne thérapeutique : expérience monocentrique
M. PENEL-PAGE*1, A. Plesa2, S. Girard3, A. Marceau-Renaut4, C. Renard1, Y. Bertrand1
1 Hématologie Pédiatrique, Institut d’Hématologie et d’Oncologie Pédiatrique, LYON ; 2 Laboratoire d’hématologie, Hôpital de Lyon Sud, Hospices Civils de Lyon et CRCL, INSERM 1052-CNRS 5286, Lyon ; 3 Service d’hématologie biologique, Centre de Biologie Est, Hospices civils de Lyon, Lyon ; 4 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille
Introduction. Les leucémies aiguës myéloblastiques (LAM) de l’enfant sont associées à une survie à long terme de seulement 50 %. Dix pour cent des patients présentent une maladie réfractaire à la première ligne de traitement, avec le protocole ELAM02 utilisé jusqu’en 2018. Il n’y a pas de recommandation concernant les traitements de seconde ligne pour ces patients. Le Gemtuzumab ozogamicine (GO), anticorps monoclonal anti-CD33 couplé à un agent cytotoxique, a montré un bénéfice dans les LAM de l’enfant et de l’adulte. Nous rapportons ici l’efficacité et la tolérance d’un traitement de rattrapage par fludarabine, cytarabine et GO (FLA-GO), pour les patients en échec de première ligne.
Patients et méthodes. Huit patients (sex-ratio H/F = 1), d’âge médian 14,5 ans (11,1-17,5), ont été traités entre 2013 et 2018 dans notre centre, après échec d’induction et de première consolidation selon le protocole ELAM02. La blastose médullaire médiane était de 30 % (2-70). Tous ont reçu une cure de FLA-GO, avec cytarabine 2000 mg/m2/j sur 3 heures de J1 à J5, fludarabine 30 mg/m2/j sur 30 minutes de J1 à J5, et 3 injection de GO 3 mg/m2 sur 2 heures à J1, J4 et J7.
Résultats. Cette cure a permis d’obtenir une rémission complète (RC) avec maladie résiduelle < 10^-2 pour 6 des 8 patients, dont 5 ont reçu une deuxième cure en consolidation avant allogreffe (avec seulement 1 ou 2 injections de GO). Le traitement a été un échec pour les 2 autres patients.
Sur 13 cures effectuées, nous avons observé une seule réaction allergique modérée, et 11 neutropénie fébriles, dont 6 avec septicémie non compliquée. Il n’y a pas eu d’infection fongique, ni de maladie veino-occlusive (MVO) post-GO. Tous les patients ont eu une toxicité hématologique de grade IV, avec une sortie d’aplasie et une reconstitution plaquettaire (100 G/L) en médiane à J34. Deux patients ont présenté une reconstitution hématologique absente ou partielle, avec des cytopénies persistantes jusqu’au début du conditionnement (J70), malgré G-CSF.
Tous les patients ont bénéficié d’une allogreffe de cellules souches hématopoïétiques après conditionnement myéloablatif, dont 7 patients en situation de RC. Cinq ont reçu du défibrotide en préventif de J1 à J21 post-greffe, dont 3 ont présenté une MVO légère à modérée. Un patient a présenté une hémorragie cérébrale sous défibrotide. Trois patients n’ont pas reçu de défibrotide préventif : deux ont présenté une MVO modérée à sévère, dont l’un a nécessité 10 jours d’hémodialyse.
Deux patients (dont l’un réfractaire au FLA-GO) ont rechuté à 1 et 4 mois post-greffe et sont décédés de l’évolution de leur LAM. Six patients sont en rémission complète persistante, avec un recul médian de 41 mois (11-51).
Conclusion. Le traitement par FLA-GO est efficace en rattrapage des LAM réfractaires à la première ligne thérapeutique, avec une tolérance acceptable. L’allogreffe de cellules souches hématopoïétique reste nécessaire afin d’obtenir une rémission prolongée. Nos données sont en faveur de l’utilisation de défibrotide préventif lors de l’allogreffe pour les patients ayant reçu une dose cumulée d’au moins 9 mg/m2 de GO.
06-20
Les leucémies aiguës myéloïdes : aspects épidémiologiques, cliniques, biologiques et thérapeutiques : rapport du groupe algérien de travail des leucémies aiguës myéloïdes/syndromes myélodysplasiques (période 2011-2017)
A. Bekadja*1, MA Bekadja coordinateur GAT LAM/SMD
1 Hématologie et Thérapie Cellulaire, Établissement Hospitalier Universitaire 1er-Novembre, Oran, Algérie
Introduction. Les LAM, ont fait l’objet de deux enquêtes nationales antérieures (M. Benakli1, MA. Bekadja2), sur deux périodes différentes (1995-2005 et 2006-2010). Cette troisième enquête nationale a pour objectifs d’actualiser les données précédentes, mais également d’en évaluer les résultats par rapport à ceux des deux périodes antérieures.
Patients et méthodes. Il s’agit d’une étude rétrospective, multicentrique, nationale, portant sur une période de 7 ans (2011-2017), incluant les patients âgés de plus de 15 ans et atteins d’une LAM primitive ou secondaire. Une fiche technique, renfermant les items anthropologiques, les signes cliniques et biologiques au diagnostic, et les moyens thérapeutiques, a été diffusée à tous les services d’Hématologie (adultes) du pays, au nombre de 19. Les incidences ont été calculées en taux pour 100 000 personnes-années et les taux standardisés par âge, en utilisant les données en population par âge et sexe de l’office national des statistiques, actualisées à l’année 2017, à l’aide du logiciel SPSS version 20.
Résultats.
Conclusion. Cette troisième étude, montre une augmentation de l’incidence annuelle des LAM (0,53 vs 0,9 vs 1,2). Les traitements d’induction doivent être homogénéisés. Par ailleurs des efforts doivent être réalisés en matière de réanimation hématologique (17 % de décès en induction). En sachant que ¾ des pts sont âgés < 60 ans, l’allogreffe de CSH doit être plus développée.
06-21
Étude du statut mutationnel de FLT3 au cours des leucémies aiguës myéloïdes : expérience de l’EHU Oran
H. Ouldjeriouat1, A. Krim2, MA. Mazari2, B. Entasoltan1, N. Yafour1, F. Serradj2, W. Boughrara3, M. Aberkane3, A. Bekadja*1
1 Hématologie et thérapie cellulaire, Établissement Hospitalier Universitaire 1er-Novembre, Oran, Algérie ; 2 Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie ; 3 Service de cytogénétique, Établissement Hospitalier Universitaire 1er-Novembre, Oran, Algérie
Introduction. Le fms-like tyrosine kinase-3 (FLT3) est un récepteur de tyrosine kinase qui joue un rôle déterminant dans la survie cellulaire, la prolifération et la différenciation des cellules souches hématopoïétiques. La mutation de FLT3 a été décrite pour la première fois en 1997. Les mutations du gène codant pour la protéine fms-like tyrosine kinase 3 (FLT3) sont les événements génétiques récurrents les plus représentés dans les leucémies aiguës myéloïdes (LAM) (FLT3) étant retrouvés dans environ 30 % des cas. Il existe deux catégories de mutations FLT3 : la duplication interne en tandem (ITD) dans le domaine juxtamembranaire du récepteur (FLT3-ITD, environ 25 %) et des mutations ponctuelles entraînant des substitutions d’acides aminés uniques dans la boucle d’activation du domaine tyrosine kinase (TKD) ou (FLT3-TKD, environ 5 %-7 %). En comparaison avec les patients atteints de LAM avec FLT3 sauvage, la présence d’une mutation ITD confère un pronostic péjoratif avec un risque de rechute élevée.
À partir de 2014, et en collaboration avec le service de cytogénétique et de biologie moléculaire, nous avons introduit la recherche du statut mutationnel de FLT3 dans le pronostic des LAM. Le but de ce travail, est d’évaluer le statut mutationnel de FLT3 au diagnostic et son impact sur la réponse thérapeutique, la rechute et la survie.
Patients et méthodes. À la date du 31 décembre 2017, 75 patients, d’âge > 15 ans, atteints de LAM (sauf LAM3) dont 41 hommes et 34 femmes avec un sex-ratio de 1,2 et ayant fait l’objet d’une recherche du statut mutationnel de FLT3, ont été colligés. Le diagnostic a été porté par cytologie, cytochimie et CMF. La recherche de la mutation de FLT3 a été réalisée par PCR. Tous les pts ont reçu une induction de type 3+7 (60/100), avec en cas de RC, suivie de cycles de consolidation avec de l’Arac HD (2 g/m2 J1, J3, J5) et ± allogreffe de CSH. L’évaluation a porté sur les taux de réponse (RC), les taux de rechute et de survie par la méthode de Kaplan-Meier. La date de point de l’étude est le 30 mai 2018.
Résultats. Parmi les 75 pts ayant fait l’objet d’une recherche de FLT3, 66 pts ont présentés un gène FLT3 sauvage (G1) avec un âge médian de 37,5 ans (16-63) dont 35 H et 31 F, sr = 1,12. Les taux moyens de GB, d’Hb, de plaquettes, de blastose périphérique et médullaire sont respectivement de 10 G/L, 8 g/dL, 74 G/L, 56 % et 64 %. 9 pts (12 %) ont présenté une mutation de type FLT3-ITD (G2), d’âge médian de 30 ans (18-57), hommes = 6, femmes = 3, sr = 2. Les taux moyens de GB, d’Hb, de plaquettes, de blastose périphérique et médullaire sont respectivement de 54 G/L, 7 g/dL, 36 G/L, 68 % et 69 %. En termes de réponse, on note 45 RC (75 %) parmi les pts évaluables, dans G1 et 6 RC (67 %) dans G2 (p = 0,7), 18 rechutes (40 %) dans G1 et 4 rechutes (67 %) dans G2 (p = 0,2). La médiane de survie globale est de 30 mois dans G2 et 10 mois dans G1 (p = 0,01).
Conclusion. La fréquence de la mutation FLT3-ITD dans notre série est faible par rapport à la littérature (12 % vs 25-30 %), par contre son caractère péjoratif est retrouvé, avec une forte hyperleucocytose, 54 G/L vs 10 G/L, un taux de blastes élevé, 68 % vs 56 %, un taux de rechute élevé (67 % vs 40 %) et une médiane de SG faible, 10 mois (p = 0,031). Ce travail doit être poursuivi pour inclure un nombre de pts plus important et valider ces résultats préliminaires.
06-22
Les expressions antigéniques dans les leucémies aiguës myéloïdes de l’adulte : expérience de l’EHU 1er-Novembre
B. Mansour1, M. Brahimi1, A. Krim2, N. Yafour1, F. Serradj2, MA. Mazari2, H. Ouldjeriouat1, B. Entasoltan1, S. Osmani1, R. Bouhass1, A. Arabi1, A. Bekadja*1
1 Hématologie et thérapie cellulaire, Établissement Hospitalier Universitaire 1er-Novembre, Oran, Algérie ; 2 Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie
Introduction. La cytométrie en flux constitue un des examens clés dans la classification des leucémies aiguës. Le but de notre travail est d’identifier le profil immunophénotypique des LAM et d’évaluer la fréquence des marqueurs aberrants.
Patients et méthodes. Il s’agit d’une étude rétrospective de juin 2009 à mai 2018 portant sur les patients présentant une LAM au diagnostic et ayant bénéficié d’un immunophénotypage par CMF selon la classification de l’EGIL. L’acquisition est réalisée sur un cytomètre en flux de type Facs Cantoll à 8 couleurs. Les critères diagnostiques de LAM retenus, sont ceux de la classification EGIL des LAM :
– positivité d’au moins deux marqueurs myéloïdes CD13/CD33/CD117/CyMPO.
– négativité d’au moins trois des marqueurs lymphoïdes T CyCD3/CD2/CD5/CD7/CD8.
– négativité d’au moins deux des marqueurs lymphoïdes B CD19/CyCD22/CyCD79a.
Résultats. Sur une période allant de juin 2009 à mai 2018, 208 leucémies aiguës ont été répertoriées. Leurs répartitions selon la classification OMS, est la suivante : 57 LAL, 4 LA biphénotypiques, 1 LA indifférenciée et 190 LAM. La classification FAB est la suivante : M0 (10 %), M1 (37 %), M2 (19 %), M3 (7 %), M4 (21 %), M5 (23 %), M6 (6 %), M7 (1 %). Les résultats de la CMF, sont les suivants : les marqueurs CD13, CD33 et CD117 sont positifs dans 83 %, 81 % et 59 % respectivement et 37 % des LAM sont CD13+/CD33 +/CD117+. La MPO est positive dans 59 % des LAM. Les marqueurs T : le CD7 et le CD3 sont positifs dans 3,7 % et 0 % respectivement. Les marqueurs B : le CD19, CD22 et le CD79a sont positifs dans 5,5 % ; 11 % et 2 % respectivement.
Conclusion. Dans notre série de patients, le CD117 est le marqueur le moins sensible mais le plus spécifique pour le diagnostic des LAM, vu que ce dernier n’a jamais été retrouvé dans les LAL (Brahimi M et al. CNH, 2015). L’utilisation de la combinaison CD13/CD33/CD117 et MPO a permis de classer 100 % des leucémies aiguës en LAM. Les marqueurs aberrants ont été retrouvés dans 16 % des cas de LAM vs 25 % et 55 % des LAL B et T respectivement dans une étude antérieure (Brahimi M et al. CNH 2015).
06-23
Colonisation et infection à l’entérocoque résistant à la vancomycine chez les patients atteints d’une leucémie myéloïde aiguë nouvellement diagnostiquée au service d’hématologie et d’oncologie pédiatrique Casablanca
W. Matrane*1, M. Lamchahab2, S. Laajouri3, N. Khoubila3, S. Cherkaoui3, M. Qachouh1, M. Bendari3, M. Rachid4, M. Harif3, A. Madani3, A. Quessar5
1 Hématologie clinique et oncologie pédiatrique, Hôpital 20-Août, Casablanca, Maroc ; 2 Hématologie et oncologie pédiatrique, CHU Ibn Rochd, Casablanca, Maroc ; 3 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc ; 4 Service d’hématologie clinique et oncologie pédiatrique, centre hospitalier universitaire Inb Rochd, Casablanca, Maroc ; 5 Hématologie et Oncologie Pédiatrique -CHU Ibn Rochd, Faculté de médecine et de pharmacie -Casablanca, Casablanca, Maroc
Introduction. Bien que L’entérocoque résistant à la vancomycine (ERV) soit réputé peu pathogène, il peut donner des infections sévères, particulièrement difficiles à traiter chez l’immunodéprimé. La colonisation intestinale joue un rôle clé dans son épidémiologie et sa pathogénie. En Amérique du Nord, les entérocoques sont observés dans environ 10 % d’hémocultures. En Europe, la fréquence de ces isolements invasifs serait plus faible (7 %) ainsi qu’en Amérique du Sud (4 %). La prévention de la diffusion repose sur l’identification précoce et l’isolement des patients porteurs et le traitement dépend de la souche en cause et de sa sensibilité antibiotique.
Patients et méthodes. Il s’agit d’une étude rétrospective étalée de janvier 2014 au décembre 2017. Tous les patients suivis pour LAM de tout âge ont été inclus. Nos patients sont hospitalisés dans une unité protégée, dans des salles individuelles à écoulement laminaire d’air filtré. Les patients portaient des cathéters veineux centraux et/ou KTP et parfois sonde urinaire. Des cultures d’écouvillonnage rectal et nasal pour l’ERV et d’autres germes ont été obtenues systématiquement à l’admission et une fois/semaine par la suite dans le cadre de nos mesures de contrôle de l’infection.
Résultats. Dans notre série, 67 cas d’ERV ont été identifiés. La médiane d’âge des patients était de 47 ans, avec un sex-ratio à 1. 82 % (n = 55) des cas correspondaient à des écouvillons rectaux positifs et 18 % (n = 12) correspondaient à des échantillons cliniques positifs. La durée médiane d’hospitalisation était de 31 jours (10-47 J) avec une durée médiane de colonisation de 10,5 semaines. Concernant les patients colonisés, 20 % étaient porteurs d’ERV au moment de leur premier examen, le reste l’a contracté pendant leur hospitalisation. Les décès ont été notés chez 27,3 % de ces patients, dont 60 % des cas étaient dus à leur maladie oncologique et 40 % étaient secondaires à des sepsis sans preuve d’ERV dans le sang. La prise des glycopeptides a été signalée chez 69 % des patients colonisés. Parmi les infections observées, 83,3 % étaient des bactériémies et 16,7 % étaient des infections urinaires. Une colonisation préalable a été établie chez quatre patients et la prise de Vancomycine a été signalée chez cinq patients infectés. Six patients infectés ont survécu à leurs épisodes neutropéniques. Un décès par sepsis grave à l’ERV a été signalé chez un seul patient.
Conclusion. Nos résultats montrent que le risque d’acquisition d’ERV est directement proportionnel à la durée du séjour à l’hôpital. Ainsi l’apparition d’ERV dans un établissement hospitalier doit conduire à une intervention rapide et énergique. Faute de quoi, la dissémination se fait à bas bruit, rapidement et à grande échelle.
06-24
L’inhibition de l’aldéhyde déshydrogénase élimine les cellules souches de la leucémie tout en épargnant les progéniteurs normaux
J. Colle1, I. Arnoux2, G. Venton1, C. Baier3, Y. Labiad3, G. Martin4, I. Ceylan4, L. Farnault1, R. Costello*1
1 Hématologie, Assistance Publique des Hôpitaux de Marseille, Université de la Méditerranée, Marseille ; 2 Laboratoire d’hématologie, Hôpital de la Timone, Marseille ; 3 Tagc, Aix-Marseille Université, INSERM, Marseille ; 4 Recherche et développement, AdvancedBioDesign, Lyon
Introduction. Les cellules souches de la leucémie (CSL) jouent un rôle important dans l’initiation, la progression et la rechute de la leucémie et représentent donc une cible critique pour une intervention thérapeutique. Cependant, peu d’agents ciblent les CSL, ce qui ralenti les progrès dans le traitement de la leucémie myéloïde aiguë (LAM). Les aldéhydes déhydrogénase sont des marqueurs des CSL et leur inhibition est une voie thérapeutique innovante dans les LAM (1,2). Nous présentons ici une étude prospective visant à définir quels patients sont susceptibles de bénéficier du traitement par DIMATE.
Patients et méthodes. Une série de 140 échantillons de LAM en première, deuxième et troisième lignes ont été analysés en cytométrie de flux pour leurs niveaux de ROS, d’aldéhyde déshydrogénase et de glutathion.
Résultats. Des études mécanistiques ont révélé que la mort cellulaire induite par le DIMATE était liée à une série d’événements métaboliques, notamment la génération d’aldéhyde toxique (HNE et MDA), la déplétion en GSH, la production excessive de ROS et l’induction de gènes associés à la réponse au stress et à l’apoptose. Notre analyse des ROS, GSH et ALDH sur échantillons de patients atteint de LAM a révélé qu’il s’agissait de biomarqueurs pronostiques et prédictifs, significativement corrélés aux facteurs pronostiques existants mais aussi à l’efficacité in vitro du DIMATE.
Conclusion. Sur la base de ces résultats, nous proposons que le DIMATE soit un agent puissant qui cible sélectivement les CSL et pourrait être utile dans le traitement de la LAM.
06-25
Étude du profil moléculaire des leucémies aiguës myéloïdes de l’adulte jeune : vers une approche thérapeutique personnalisée ? Résultats de l’étude ALFA-0702
L. Fenwarth*1, R. Itzykson2, N. Duployez1, C. Berthon3, P. Sujobert4, K. Celli-Lebras5, A. Marceau-Renaut1, A. Pigneux6, S. De Botton7, S. Chantepie8, N. Vey9, E. Raffoux10, D. Caillot11, P. Turlure12, E. Lemasle13, C. Pautas14, S. Chevret15, X. Thomas16, H. Dombret10, C. Preudhomme1
1 Laboratoire d’hématologie, CHU Lille, Lille ; 2 Hématologie, Hôpital Saint-Louis, Paris ; 3 Maladies du sang, CHU Lille, Lille ; 4 Hématologie, CH Lyon Sud, Pierre-Bénite ; 5 Coordination groupe alfa, Assistance Publique Hôpitaux de Paris, Paris ; 6 Hématologie, Hôpital Haut-Lévêque, Pessac ; 7 Service d’hématologie clinique, Institut Gustave Roussy, Villejuif ; 8 Service d’hématologie clinique, CHU Caen, Caen ; 9 Epigenetic factors in normal and malignant hematopoiesis, CRCM-Inserm, U1068 ; Institut Paoli-Calmettes ; Aix-Marseille Université, UM 105 ; CNRS, UMR7258, Marseille ; 10 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 11 Service d’hématologie, CHU de Dijon, Dijon ; 12 Hématologie, CHU Limoges, Limoges ; 13 Département d’hématologie, Hôpital Henri Becquerel, Rouen ; 14 Hématologie clinique et thérapie cellulaire, Hôpital Henri Mondor, Créteil ; 15 Service de biostatistique et information médicale, Hôpital Saint-Louis, Paris ; 16 Hématologie, Centre Hospitalier Lyon Sud, Lyon
Introduction. Les LAM représentent des entités hétérogènes sur le plan clinique et biologique. L’étude des altérations génétiques des LAM permet de stratifier ces patients selon leur profil mutationnel dans la dernière classification ELN 2017. Cette classification ne tient compte que d’un nombre restreint d’altérations cytogénétiques et moléculaires. Récemment, des modèles multifactoriels de prédiction ont été développés afin de mieux apprécier le pronostic des patients. L’objectif de ce travail était d’analyser le profil moléculaire des LAM des adultes jeunes issus du protocole ALFA-0702 (#NCT00932412) afin de valider la classification ELN 2017 et le modèle de prédiction pronostique développé par Gerstung et al. (1), récemment validé par Huet et al. dans une cohorte rétrospective monocentrique de 155 patients (2).
Patients et méthodes. Cette étude incluait les patients âgés de 18 à 59 ans présentant une LAM de novo entre septembre 2009 et mars 2013 issus du protocole ALFA-0702 pour lesquels de l’ADN au diagnostic était disponible pour séquençage haut débit (next generation sequencing, NGS). L’analyse NGS a été réalisée sur une librairie HaloplexTM (Agilent®) incluant 36 gènes. La comparaison des valeurs prédictives de l’ELN 2017 versus l’approche « banque de connaissance » (score de Gerstung) sur la survie globale à trois ans s’est faite par estimation et comparaison des courbes ROC temps-dépendantes pour des données censurées (2).
Résultats. L’analyse moléculaire centralisée a été réalisée chez 656 des 713 patients inclus dans l’essai ALFA-0702. 93,4 % des patients présentaient au moins une mutation. Les gènes les plus fréquemment mutés étaient NPM1 (35,7%), DNMT3A (26,8 %) FLT3-ITD (22,4 %), NRAS (22,0 %) et TET2 (13,0 %). La régulation épigénétique (mutations de TET2, IDH1, IDH2, DNMT3A, ASXL1, BCOR, BCORL1, EZH2 ou MLL-PTD, 59,0 %) et la voie de transduction du signal (mutation de FLT3-ITD, FLT3-TKD, KIT, NRAS, KRAS, CSF3R, JAK2, KIT, PTEN, PTPN11 ou RIT1, 57,4 %) représentaient les voies fonctionnelles les plus fréquemment altérées. Parmi les 42 patients avec échec de cytogénétique, 8 présentaient des mutations de TP53, ASXL1 ou RUNX1 permettant de les classer en ELN défavorable, et 42 demeuraient inclassables. Parmi les 622 patients évaluables, 173 (27,8 %), 201 (32,3 %), 248 (39,9 %) patients étaient à risque favorable, intermédiaire ou défavorable, respectivement selon la classification ELN 2017. Avec un suivi médian de 4,15 ans, la survie globale à 5 ans (sans censure à l’allogreffe) était respectivement estimée à 78 % (IC95% : 70-83), 59 % (IC95% : 51-65) et 44 % (IC95% : 37-51) pour chacun de ces groupes (test du log-rank, p < 10-4). Sur les 100 variables clinicobiologiques prises en compte par l’algorithme décisionnel décrit par Gerstung et al., 4 des 16 variables cliniques et 24 des 84 variables biologiques étaient manquantes (correspondant à des gènes très rarement mutés et peu informatifs sur le plan pronostique). Ce modèle permettait de définir plus précisément la probabilité de survie globale à 3 ans que le score ELN 2017 (p < 10-8). L’évaluation de la prédiction du bénéfice de l’allogreffe est en cours d’analyse.
Conclusion. La stratification oncogénétique selon la classification ELN 2017 discrimine le pronostic des LAM de l’adulte de moins de 60 ans traitées prospectivement dans un essai clinique. Un modèle pronostique reposant sur une banque de données améliore la prédiction de la survie à trois ans.
06-26
Le nombre de mutations additionnelles hors JAK2, CALR et MPL est pronostique dans les leucémies aiguës myéloïdes postsyndrome myéloprolifératif
E. Chiche*1, S. Bonnet2, S. Bertoli3, A. Kuykendall4, L. Gastaud5, V. De Mas6, E. Delabesse7, A. Marceau-Renaut8, C. Preudhomme9, A. Murati10, D. Sallman11, S. Raynaud12, C. Récher13, N. Vey14, T. Cluzeau15
1 Hématologie Clinique, CHU de Nice, Nice ; 2 Hématologie, Institut Paoli-Calmettes, Marseille ; 3 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 4 Hématologie, Moffitt Cancer Center et Research Institute, Tampa, États-Unis ; 5 Oncologie, Centre Antoine Lacassagne, Nice ; 6 Iuct, Inserm, Toulouse ; 7 Oncopole, CHU Toulouse - Casselardit Ancely, Toulouse ; 8 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 9 Laboratoire d’hématologie, CHU Lille, Lille ; 10 Laboratoire d’hématologie, Institut Paoli-Calmettes, Marseille ; 11 Hematology, Moffitt Cancer Center, Tampa, États-Unis ; 12 CHU, Nice ; 13 Hématologie adulte, Institut Universitaire du Cancer de Toulouse-Oncopole, Toulouse ; 14 Epigenetic factors in normal and malignant hematopoiesis, CRCM-Inserm, U1068 ; Institut Paoli-Calmettes ; Aix-Marseille Université, UM 105 ; CNRS, UMR7258, Marseille ; 15 INSERM U1065, Centre Méditerranéen de Médecine Moléculaire, Nice
Introduction. Les leucémies aiguës myéloïdes (LAM) secondaires à un syndrome myéloprolifératif (SMP) surviennent dans 1,5 %, 7 % et 11 % des patients présentant respectivement une thrombocytémie essentielle (TE), une polyglobulie de Vaquez (PV) et une myélofibrose primitive (MFP). Peu de données moléculaires sont disponibles actuellement pour ce sous-groupe de très mauvais pronostic.
Matériels et méthodes. Les données cliniques et biologiques ont été analysées chez 111 patients traités pour LAM post-SMP. L’ADN à partir des prélèvements médullaires au diagnostic de SMP et de LAM a été extrait. Les mutations JAK2V617F et MPLW515L/K ont été identifiées par PCR et celles de CALR par séquençage conventionnel. Un NGS analysant 36 gènes a également été réalisé chez 96/111 patients. La réponse globale (RG) était définie par la présence d’une réponse complète (RC), d’une RC avec récupération hématologique incomplète (RCi), d’une réponse partielle (RP) ou d’une maladie stable. La survie globale (SG) a été calculée à partir de la date du diagnostic et de celle des dernières nouvelles ou du décès. Toutes les analyses statistiques ont été réalisées à partir du logiciel SPSS v.22 (IBM SPSS Statistics).
Résultats. Chez ces 111 patients, le sex-ratio H/F était de 1,17 et l’âge médian au diagnostic de LAM de 66 ans. L’analyse cytogénétique était favorable, intermédiaire et défavorable chez 2, 51 et 47 patients, respectivement. Le nombre médian de mutations additionnelles (hors JAK2, MPL ou CALR) était de 2 [0-6]. Les plus fréquentes étaient celles de TP53 (23%), ASXL1 (17%), TET2 (13%) et SRSF2 (10%). Seuls 2 patients étaient mutés pour le gène NPM1, 2 pour FLT3ITD et 4 pour FLT3TKD.
Le SMP d’origine était la PV, la TE et la MFP chez 20 %, 34 % et 46 % des patients, respectivement. Le traitement choisi en première ligne était une chimiothérapie intensive (CI) pour 61 (55 %) patients, un agent hypométhylant (HM) chez 10 (9 %) ou un autre traitement (incluant les soins de supports et la cytoréduction) pour tous les autres. 24/111 (22 %) ont bénéficié d’une allogreffe de cellule souches hématopoïétiques (CSH). Le taux de RG était de 54 % chez les patients traités par CI ou HM (avec 30/71 (42 %) en RC/RCi).
Nous n’avons pas identifié de facteur prédicteur de RC/RCi. La SG était de 12 mois [6-18] et ne dépendait ni du risque cytogénétique, ni du type ou de la fréquence allélique (VAF) des mutations JAK2/MPL/CALR, ni de la réalisation d’une allogreffe de CSH. La SG était significativement plus longue chez les patients traités par HM (46 mois [21-71] versus 10 mois [8-12] avec la CI, p = 0,006), ayant eu une PV en phase chronique (26 mois [0-57] versus 10 mois [7-13] et 10 mois [4-16] dans les LAM post TE et post MFP, respectivement, p = 0,07) et présentant au moins une mutation additionnelle (46 mois [32-60] chez 58 patients versus 5 mois [0-12] chez 38 patients sans mutations, p = 0,04). Seul ce dernier paramètre était significatif dans l’analyse multivariée sur la SG avec un hazard ratio à 0,32 [0,13-0,82] (p = 0,02).
Conclusion. Le pronostic défavorable des LAM post-SMP a pu être confirmé par cette étude. Seule la présence ou non de mutations additionnelles s’est révélée pronostique dans ce sous-groupe. Le développement d’essais cliniques spécifiques à ce sous-groupe reste encore aujourd’hui un enjeu médical.
06-27
Exploration génomique d’un déficit en myéloperoxydase dans une leucémie aiguë promyélocytaire
B. Ducourneau1, C. Roumier2, N. Helevaut3, F. Dumezy4, S. Tricot5, A. Daudignon6, N. Cambier5, N. Duployez7, C. Preudhomme7, S. Poulain*8
1 Laboratoire d’hématologie, Ch de Valenciennes et CHRU de Lille, Lille ; 2 Laboratoire d’hématologie, Centre de Biologie - Pathologie, Lille ; 3 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 4 Laboratoire d’hématologie, CH Régional Universitaire de Lille, Lille ; 5 Service d’hématologie clinique, CH De Valenciennes, Valenciennes ; 6 Laboratoire de cytogénétique Jeanne de Flandres, CHU Lille, Lille ; 7 Laboratoire d’hématologie, CHU Lille, Lille ; 8 Laboratoire d’Hématologie, CHU de Lille, Lille
Introduction. La leucémie aiguë promyélocytaire (LAP) se caractérise par une infiltration médullaire par des blastes de morphologie promyélocytaire avec des corps d’Auer en fagots et par un réarrangement PML-RARA/t (15 ; 17). Les LAP constituent un sous-groupe de LA myéloïde qui bénéficie de thérapie ciblée avec des agents différentiants. Les translocations cryptiques ou variantes sont observées dans < 5 % des cas. La myéloperoxydase (MPO) est une peroxydase héminique dont l’expression caractérise les granulations primaires des cellules myéloïdes. La cytochimie de la myéloperoxydase, classiquement positive dans les LAP, peut être utilisée en urgence pour le diagnostic différentiel dans les formes atypiques ou hypogranulaires. L’absence d’activité peroxydasique est exceptionnelle. Un cas a été associé à la présence d’un variant au niveau d’un site de splice intronique à l’état homozygote (rs 35897051) liée à une disomie uniparentale acquise en 17q [Heiblig et al., Blood 2016]. Nous rapportons l’exploration moléculaire d’un cas de LAP avec déficit en myéloperoxydase.
Patients et méthodes. Un patient de 49 ans est adressé pour une altération de l’état général. L’hémogramme montre une leuconeutropénie (GB : 0,87 × 109/L) et une thrombopénie (plaquettes : 56 × 109/L), le myélogramme une infiltration par 89 % de blastes hypergranuleux d’aspect promyélocytaire avec de très exceptionnels corps d’Auer en fagots. L’immunophénotypage confirme la nature myéloïde des blastes (CD13+, CD117+, CD33+ HLA-DR négatif et CD34 low). Un faible niveau d’expression de la MPO est observé. La cytochimie de la MPO, réalisée par technique à la benzidine, est négative dans les blastes. Le caryotype conventionnel met en évidence une trisomie 8. La réalisation de la FISH PML RARA permet d’établir la formule chromosomique : 47, XY, +8[2]/46,XY[20].ish ins (15;17) (q22;q21q21)[4]. Le transcript PML-RARA bcr1 est identifié en RT-PCR. Le diagnostic de LAP est donc posé. Le patient est traité par acide tout-trans rétinoïque associé à de l’idarubicine et de cytarabine. Une rémission complète (RC) est observée.
Résultats. Afin de comprendre le mécanisme moléculaire associé à cette présentation atypique de LAP, le gène codant la MPO, localisé en 17q 22 et comprenant 12 exons, a été séquencé par technique Sanger. Une mutation monoallélique dans l’exon 6 (c.749T>C), MPO L250P, est identifiée au niveau de la région codant pour le domaine peroxydase au niveau de la chaîne légère. Cette mutation est décrite comme potentiellement délétère par les logiciels d’analyse in silico et pourrait modifier l’emplacement de l’hème ainsi que le repliement, la fonction et la stabilité de la MPO. Cette mutation est acquise et n’a jamais été décrite dans les déficits constitutionnels en MPO. Le rs35897051 est observé à l’état hétérozygote au diagnostic et à la RC. La SNP array ne révèle pas de disomie uniparentale anomalie en 17q.
Conclusion. Les LAP avec déficit d’activité en myéloperoxydase sont des entités exceptionnelles. Nous rapportons un nouveau mécanisme moléculaire de perte d’activité peroxydasique dans les LAP par mutation acquise du gène de la MPO associé à une diminution d’expression de l’antigène dans les blastes.
06-28
Suivi de la maladie résiduelle par quantification des mutations rares de NPM1 par PCR digitale dans les leucémies aiguës myéloïdes
A. Lesieur*1, O. Nibourel1, A. Marceau1, L. Fenwarth1, C. Frimat1, C. Rodriguez1, K. Celli-Lebras2, B. Quesnel3, D. Lebon4, N. Boissel5, S. Castaigne6, X. Thomas7, H. Dombret8, C. Preudhomme1, N. Duployez1
1 Centre de biologie pathologie - laboratoire hématologie, CHU Lille, Lille ; 2 Coordination groupe alfa, Hôpital Saint-Louis, AP-HP, Paris ; 3 Département d’hématologie, CHU Lille, Lille ; 4 Département d’hématologie, CHU Amiens, Amiens ; 5 Département d’hématologie, Hôpital Saint-Louis, AP-HP, Paris ; 6 Département d’hématologie, CH Versailles, Le Chesnay ; 7 Département d’hématologie, CH Lyon Sud, Lyon ; 8 Département d’hématologie, Hôpital Saint-Louis, AP-HP, Paris
Introduction. Les mutations du gène NPM1 sont retrouvées dans 35 % des LAM, et dans 60 % des LAM à caryotype normal pour lesquelles elles constituent un facteur pronostique majeur [1]. Plus de 50 mutations localisées dans l’exon 11 du gène NPM1 ont été identifiées [2]. En pratique clinique la quantification des transcrits NPM1-mutés est réalisée par RT-PCR quantitative en temps réel (qPCR) pour les formes A, B et D qui prédominent chez 95 % des patients. Cette quantification n’est pas réalisée pour les autres variants, dits rares, à défaut de calibrant existant. L’objectif de cette étude est d’évaluer l’aptitude de la PCR digitale à quantifier les mutations rares de NPM1 afin de valider cette technique pour une utilisation prospective.
Patients et méthodes. 40 patients atteints de LAM avec une mutation rare de NPM1 ont été répertoriés pour l’étude, parmi lesquels 5 ont rechuté. Au total 18 types de variants rares de NPM1 ont été analysés sur des échantillons de sang périphérique ou de moelle osseuse, au diagnostic et en post-induction. Pour les patients ayant rechuté l’ensemble des points de suivi prélevés sur sang périphérique a également été analysé. La PCR digitale permet une quantification absolue et hautement précise de séquences d’acides nucléiques cibles par fractionnement du milieu réactionnel puis discrimination et dénombrement des fractions positives ou négatives. Dans le système développé par Bio-Rad le fractionnement est effectué par émulsion et la détection de l’amplification réalisée à l’aide de sondes à hydrolyse fluorescentes. La lecture de la fluorescence a lieu en point final. Dans notre étude, la PCR est réalisée en multiplex sur ADN complémentaire en utilisant simultanément un système amorce-sonde spécifique du transcrit NPM1-muté associé à un système spécifique du gène de référence ABL. Les performances de la méthode ont été évaluées sur la forme classique A.
Résultats. La mise au point de la technique a permis de fixer la température optimale d’hybridation des sondes à 53 ̊C pour l’ensemble des variants. Une très bonne discrimination entre les clusters positifs et négatifs a été observée. Les limites de blanc ont été déterminées par la quantification de 8 témoins négatifs. La limite de linéarité a été mesurée grâce à la quantification de 6 dilutions en série d’un plasmide commercial NPM1 de type A. Les essais de répétabilité ont montré des résultats très précis avec des duplicates toujours très proches, contrairement à la qPCR. La robustesse de la technique sera appréciée suite à l’étude de la reproductibilité actuellement en cours. Les premiers résultats de comparaison à la qPCR sont prometteurs. Les corrélations avec les données cliniques et pronostiques seront présentées lors du congrès.
Discussion. La PCR digitale est très souple d’utilisation et cette application offre plusieurs avantages par rapport à la qPCR. La quantification simultanée et dans le même puits, du transcrit muté et du gène ABL, autorise un débit élevé et permet de s’affranchir des aléas de pipetage. De plus, la quantification directe sans extrapolation à partir d’une courbe de calibration offre également une meilleure précision. Enfin, l’absence de calibrant permet de réduire le coût analytique et d’analyser des séries avec un nombre d’échantillons variables.
Conclusion. Moins contraignante et plus précise, la PCR digitale se présente comme une alternative à la qPCR pour le suivi des patients NPM1-mutés. Adaptée à la quantification des variants rares de NPM1, cette technique permettra de suivre l’évolution de la maladie résiduelle et de détecter précocement une rechute chez ces patients.
06-29
Leucémie aiguë promyélocytaire/expérience du service d’hématologie et de thérapie cellulaire de l’EHU 1er-Novembre d’Oran
F. Serradj*, A. Krim, N. Yafour, H. Ouldjeriouet, MA. Mazari, N. Hakiki, M. Brahimi, A. Bekadja
Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie
Introduction. La leucémie aiguë à promyélocytes (LAP) est une entité particulière, rare, des leucémies aiguës non lymphoblastiques, caractérisée par une prolifération des myélocytes et des promyélocytes anormaux. Le caryotype objective une translocation t(15;17)(q22;q21) et la PCR détecte le transcrit de fusion PML-RARα, qui entrave la fonction du produit normal de PML et inhibe la maturation des promyélocytes. L’utilisation de l’acide tout transrétinoïque (ATRA) a complètement changé le pronostic des leucémies à promyélocytes.
Patients et méthodes. Il s’agit d’une étude monocentrique, rétrospective, portant sur une période allant de juin 2009 à décembre 2017, et réalisée à partir de dossiers médicaux de patients, âgés de plus 15 ans et présentant une LAP. Le diagnostic a été posé sur la cytochimie au Noir Soudan, la cytométrie en flux et la cytogénétique à la recherche de la translocation t(15;17). La biologie moléculaire à la recherche du transcrit de fusion PML-RARα n’a pu être réalisée. La recherche de groupes pronostics a été réalisée selon la méthode du score de Sanz. Tous les patients ont reçu un même traitement d’induction de type « 3+7 » (daunorubicine 60 mg/m2/cytarabine 200 mg/m2) +ATRA per os, 45 mg/j avec consolidation si RC, selon le protocole APL2000. Un traitement d’entretien a été institué en associant méthotrexate à 15 mg/m2/semaine+Purinéthol 90 mg/m2/j+ATRA 45 mg/m2/j pendant 15 jours tous les 3 mois pendant 2 ans. L’évaluation a porté sur les réponses, la survie globale et la survie sans rechute à l’aide du Logiciel SPSS version 20.
Résultats. Sur une période de 9 ans, 197 patients atteints de LAM ont été colligés et parmi eux 12 patients (6 %) atteints de LAP ont été recensés, dont 7 femmes (sex-ratio = 0,58) avec un âge médian de 38 ans (15-62 ans). Sur le plan clinique, on note un syndrome anémique chez 9 pts (75 %), un syndrome hémorragique chez 7 pts (58 %), un patient a présenté un syndrome infectieux et un autre a présenté un syndrome tumoral. 4 (33 %) pts ont présenté un PS > 2. Du point de vue biologique, une hyperleucocytose a été notée chez 4 (33 %) pts, une pancytopénie chez 6 (50 %) pts et une CIVD compensée chez 2 (17 %) pts. Le taux de GB médian était de 1,56 G/L (0,2-36,89), celui des plaquettes de 24 G/L (4-90) et le taux d’Hb médian était de 7 g/dL (5-9). La blastose médullaire moyenne était de 72 %. Selon le score de Sanz : 6 (50 %) pts étaient à haut risque, 5 (42 %) pts, de risque intermédiaire et 1 pt de faible risque. Sur le plan thérapeutique on a relevé comme complication : une toxidermie à la cytarabine chez 2 pts, un ATRA syndrome chez 3 pts ayant bien évolué sous dexaméthasone et 3 décès précoces < 2 mois, liés à une CIVD et une hémorragie cérébrale. L’évaluation de la réponse au TRT parmi les 9 patients évaluables, a montré une RC1 chez tous les 9 (100 %) pts. Sur le plan évolutif, la SG à 64 mois est de 78 % (médiane = 70 mois) et la médiane de SSR = 44 mois. Deux patients ont rechuté à 23 et 65 mois. À la date de point du mois d’avril 2018, et avec un suivi médian de 20,5 mois, 6 (50 %) pts sont vivants et 6 (50 %) pts au total, sont décédés, des suites d’une CIVD et d’une hémorragie cérébrale chez les 3 décès précoces, un hématome sur grêle, une insuffisance rénale aiguë, une insuffisance cardiaque chez les 3 autres restants dont 2 pts en RC1 à 3 mois et à 44 mois.
Conclusion. La LAP est une hémopathie maligne rare dans notre recrutement (6 %), cependant malgré un taux élevé de RC, le taux de décès demeure élevé au cours de l’évolution.
06-30
Évaluation du protocole d’induction « 3+7 » des leucémies aiguës myéloblastiques dans la vraie vie : expérience multicentrique de l’Ouest algérien
A. Krim*1, MA. Bekadja1, B. Benzineb2, N. Mesli2, S. Zouani3, D. Saidi3, H. Touhami3, M. Aberkane4, A. Bachiri4, M. Benlazar5, Z. Zouaoui5, N. Mehalhal6, H. Gaid6, M. Talbi7, MR. Dahmane8
1 Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie ; 2 Hématologie, CHU Dr Tidjani Damardji, Tlemcen, Algérie ; 3 Hématologie, CH et Universitaire d’Oran, Oran, Algérie ; 4 Hématologie, Hôpital Militaire Régional Universitaire d’Oran, Oran, Algérie ; 5 Hématologie, CHU Hassani Abdelkader, Sidi Bel Abbès, Algérie ; 6 Hématologie, EPH Mascara, Mascara, Algérie ; 7 Hématologie, EPH Béchar, Béchar, Algérie ; 8 hématologie, hôpital Ahmed Medeghri, Saïda, Algérie
Introduction. Les leucémies aiguës occupent le troisième rang des hémopathies malignes en Algérie. Les leucémies aiguës myéloblastiques (LAM) sont les plus fréquentes avec une incidence évaluée en 2017 à 1,5/100 000 habitants. L’induction « 3+7 » représente le gold standard du traitement du sujet jeune ≤ 60 ans depuis plus de 40 ans. L’objectif de ce travail est d’évaluer les résultats thérapeutiques du protocole « 3+7 » en terme de rémission complète (RC), survie globale (SG), survie sans événement (SSE) dans la vraie vie.
Patients et méthodes. Il s’agit d’une étude régionale, multicentrique, descriptive, rétrospective portant sur une période de 2007 à 2017 au cours de laquelle tous les pts âgés entre 16 et 60 ans, atteints de LAM (LAM3 exclue) et traités par le protocole « 3+7 » ont été inclus. Le protocole « 3+7 » : daunorubicine 60 mg/m2, 90 mg/m2, 45 mg/m2 ou doxorubicine 35 mg/m2 pendant 3 jours et la cytarabine 100 mg/m2 pendant 7 jours. Les critères de réponses (RC, RP, échec) et les définitions des survies ont été calculés selon les recommandations ELN 2017. Les survies (SSE, SG) ont été calculées selon la méthode de Kaplan et Meier avec le logiciel SPSS version 20. La date de point de l’étude : 31 décembre 2017.
Résultats. 436 pts atteints de LAM colligés dont 327 pts traités par le protocole « 3+7 ». Age médian 38 ans, sex-ratio 1,4. Au diagnostic, le taux médian de GB = 18 300/μl (200-500 000) avec 14 % de pts ayant un taux > 100 000/μL. Le taux médian d’Hb = 8 g/dL (2-14), le taux médian de plaquettes = 43 000/μl (0-559 000), les taux médians de blastes périphériques et médullaires sont respectivement de 49 % (0-100) et de 75 % (12-100). 26 % des LAM sont tumorales avec des adénopathies dans 49 % des cas, une splénomégalie dans 46 %. 31 % des patients ont présenté un syndrome hémorragique, 11 % une anémie sévère et 34 % un syndrome infectieux. Classification FAB : M2 = 25%. 53 pts (16 %) ont bénéficié d’une étude cytogénétique. Selon la classification cytogénétique MRC : 7 % = pronostic favorable, 62 % = intermédiaire et 31 % = défavorable. La mutation FLT3-ITD a été recherchée chez 70 pts (21 %) ; positive chez 7 pts. Les résultats thérapeutiques : RC = 69%. Le taux de décès en induction = 12%. La SSE médiane = 11 mois avec une SSE à 2 ans = 32%. La SG médiane = 16 mois avec une SG à 2 ans = 40%. La comparaison entre les deux doses de daunorubicine 60 mg/m2 (DNR60) et 90 mg/m2 (DNR90) ne retrouve pas de différence significative en terme de RC ni de mortalité en induction ; la SSE médiane = 9 mois et 16 mois (p = 0,02) pour DNR60 et DNR90 respectivement. La SG médiane = 13 mois et 28 mois (p = 0,02) respectivement.
Conclusion. Nos résultats reflètent la prise en charge globale des LAM dans la « vraie vie ». Le taux de rémission est satisfaisant. Un effort est à entreprendre pour améliorer les taux de survies. L’intensification des doses de daunorubicine semble avoir un impact sur les taux de SSE et SG dans notre étude.
06-31
Azacitidine dans le traitement des leucémies aiguës myéloblastiques réfractaires : expérience du service d’hématologie et thérapie cellulaire de l’EHU 1er-Novembre d’Oran, Algérie
A. Krim*, F. Serradj, MA. Mazari, N. Yafour, N. Hakiki, B. Entasoltan, B. Mansour, S. Osmani, A. Arabi, M. Brahimi, R. Bouhass, MA. Bekadja
Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie
Introduction. Les leucémies aiguës myéloblastiques (LAM) réfractaires représentent 20 à 30 % des LAM du sujet jeune et 40 à 50 % des LAM du sujet âgé. L’azacitidine (AZA) est un agent hypométhylant, ayant prouvé son efficacité dans les syndromes myélodysplasiques. Peu de données en littérature existent quant à son utilisation dans les LAM réfractaires. L’objectif de ce travail est d’évaluer l’efficacité en terme de réponse globale (RG) (rémission complète et rémission complète avec récupération hématologique incomplète (RC et RCi), rémission partielle (RP)) et survie globale (SG) et la sécurité d’utilisation de l’AZA au cours des LAM réfractaires.
Patients et méthodes. Il s’agit d’une étude rétrospective de février 2015 à août 2017, portant sur les LAM réfractaires et traitées par AZA (rattrapage). La LAM réfractaire est définie par l’échec à une ligne de chimiothérapie intensive chez le sujet ≥ 60 ans et à 2 lignes chez le sujet jeune < 60 ans. L’AZA est administré à la dose de 75 mg/m2 en s/c pendant 5 à 7 jours/4 à 5 semaines jusqu’à progression. Les pts RC, RCi avec un donneur HLA compatible bénéficie d’une allogreffe géno-identique. Les critères de réponse sont définis selon l’IWG 2006. La SG est calculée de J1 de l’azacitidine jusqu’au décès, avec le logiciel SPSS version 20. La date de point de l’étude : 1er septembre 2018.
Résultats. 11 pts inclus dont 7 hommes. L’âge médian 40 ans [21-61]. À l’initiation de l’AZA : 18 % de pts avaient un taux de GB > 15 000/mm3, les taux médians de blastes périphériques et médullaires sont respectivement de 20 % (0-55) et de 30 % (8-98). 10 pts (91 %) ont bénéficié d’une étude cytogénétique. 4 pts ont un caryotype normal et 3 pts un caryotype complexe, 1 pt avec del(7), 1 pt avec t(11;19) sans remaniement MLL, 1 pt avec trisomie 13. Selon la classification MRC : 60 % intermédiaire et 40 % défavorable. La mutation FLT3-ITD a été retrouvée chez 2 pts (18 %). Le nombre médian de lignes thérapeutiques avant azacitidine = 2 [1-5]. Nombre de cycle médian d’Aza reçus = 11[3-35]. RG = 45% (18% de RC, 9 % de RCi et 18 % de RP) ; SD = 45%. Toxicité hématologique grade 3 à 4 = 55%. 2 hospitalisations pour neutropénie fébrile sur 104 cycles d’AZA. 1 pt a un donneur HLA compatible et qui a bénéficié d’une allogreffe géno-identique après 7 cycles d’AZA. La SG médiane = 12 mois.
Conclusion. L’AZA est une option thérapeutique intéressante dans les LAM réfractaires. Dans notre expérience, le taux RG est satisfaisant 45 % avec une médiane de survie de 12 mois pour des LAM dont le pronostic est très pauvre. L’allogreffe reste le traitement de choix pour les pts en RC pour améliorer leur survie.
06-32
Daunorubicine 90 mg/m2versus 60 mg/m2 dans le traitement d’induction des leucémies aiguës myéloblastiques du sujet jeune : expérience du service d’hématologie et de thérapie cellulaire de l’EHU 1er-Novembre, Oran, Algérie
A. Krim*, F. Serradj, MA. Mazari, N. Yafour, N. Hakiki, B. Entasoltan, S. Osmani, B. Mansour, A. Arabi, M. Brahimi, R. Bouhass, MA. Bekadja
Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie
Introduction. Le traitement des leucémies aiguës myéloblastiques (LAM) repose, chez les sujets jeunes ≤ 60 ans sur une induction standard « 3+7 » associant la daunorubicine (DNR) à la cytarabine (ARAC). Plusieurs essais randomisés ont prouvé qu’une intensification de la dose de la DNR à 90 mg/m2 était associée à de meilleurs taux de rémission complète (RC), de survie globale sans majoration de la toxicité. Nous rapportons dans ce travail, les résultats de l’utilisation de la daunorubicine aux doses de 60 et 90 mg/m2.
Patients et méthodes. Il s’agit d’une étude rétrospective portant sur une période de 2009 à 2016, au cours de laquelle tous les patients (pts) âgés entre 16 et 60 ans, atteints de LAM ont été inclus (LAM3 exclue). Entre 2009 et 2012, tous les pts étaient traités par le protocole « 3+7 » associant DNR 60 mg/m2 pendant 3 jours et ARAC 100 mg/m2 pendant 7 jours (Bras DNR60) et de 2013 à 2016, tous les pts étaient traités par « 3+7 » avec une intensification de la dose de la DNR à 90 mg/m2 (Bras DNR90). Les pts en RC reçoivent une consolidation (3 cycles d’ARAC Haute dose) et pour ceux qui ont un donneur HLA compatible, ils reçoivent une allogreffe géno-identique après un ou deux cycles d’ARAC HD (sauf LAM CBF). L’évaluation a porté sur la RC, les survies sans rechute (SSR) et globale (SG) calculées selon la méthode de Kaplan et Meier avec le logiciel SPSS version 20. La date de point de l’étude : 31 décembre 2017.
Résultats. 117 pts adultes atteints de LAM sont colligés, d’âge médian 36 ans [16-59], dont 53 Femmes. Bras DNR60 = 55 pts et Bras DNR90 = 62 pts Les résultats thérapeutiques montrent : 64 % et 77 % de RC dans les deux groupes DNR60 et DNR90 respectivement (p = 0,15). Pas de différence de toxicité (grade 3 et 4) hématologique et non hématologique. Taux de décès en induction similaire : 14 % et 11 % dans les groupes DNR60 et DNR90 respectivement. Pas de différence significative en terme de SSR ; la médiane est de 10 mois vs 16 mois (p = 0,13) dans DNR60 vs DNR90. La SG médiane est de 8 mois (DNR60) et 19 mois (DNR90) avec une différence significative (p = 0,01).
Conclusion. Dans notre étude, une intensification de la dose de DNR à 90 mg/m2 est associée à une SG significativement prolongée par rapport à la dose standard. Pas de différence, par contre, en terme de RC ou de SSR.
06-33
Profil cytogénétique des patients atteints de leucémie aiguë myéloblastique : Expérience du service d’hématologie et thérapie cellulaire de l’EHU 1er-Novembre d’Oran, Algérie
A. Krim*, F. Serradj, MA. Mazari, N. Yafour, N. Hakiki, B. Entasoltan, S. Osmani, B. Mansour, A. Arabi, M. Brahimi, R. Bouhass, MA. Bekadja
Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie
Introduction. Les leucémies aiguës myéloblastiques (LAM) sont un groupe hétérogène d’hémopathies malignes. Plusieurs classifications pronostiques se sont succédé pour définir les différents groupes à risque. L’influence majeure de la cytogénétique conventionnelle a été établie dans de nombreuses études de grandes cohortes de patients. Ces études ont permis une stratification en groupes pronostiques : favorable, intermédiaire et défavorable. Les plus importantes étant les classifications SWOG (Southwest Oncology Group) et MRC (Medical Research Council.).
Patients et méthodes. Il s’agit d’une étude rétrospective sur une période de 4 ans (2014-2017) portant sur les patients adultes atteints de LAM ayant bénéficié d’un caryotype conventionnel associé ou non à une étude FISH. L’étude cytogénétique a été faite sur prélèvement médullaire et/ou sanguin. L’évaluation a porté sur la fréquence des anomalies cytogénétiques, la classification pronostique cytogénétique selon SWOG et son impact sur les survies sans événement (SSE) et globale (SG) calculées selon la méthode de Kaplan et Meier avec le logiciel SPSS version 20. La date de point de l’étude est : 31 décembre 2017.
Résultats. Sur une période de 4 ans, 45 patients ont bénéficié d’une étude cytogénétique. 6 échecs de culture. Un caryotype normal a été retrouvé dans 44 % des cas, un caryotype complexe (≥ 3 anomalies) dans 15 % des cas, t(11;19) sans remaniement 11q23 dans 10 % des cas, la del(7) dans 8 % des cas, la del(9) dans 5 % des cas, 5q- dans 3 % des cas, la t(8;21) et inv(16) étaient moins fréquentes (3 %). Selon la classification pronostique SWOG, le groupe favorable (Fav) représentait (6 %), le groupe intermédiaire (Int) (61 %) et défavorable (Déf) (33 %). La médiane de SSE n’est pas atteinte dans le groupe FAV, elle est de 15 mois dans le groupe Intermédiaire et de 13 mois dans le groupe défavorable, pas de différence significative entre les deux groupes Int et Def en terme de SSE. La médiane de SG n’est pas atteinte dans le groupe favorable, de 34 mois dans le groupe Intermédiaire et de 13 mois dans le groupe défavorable avec une différence significative entre les trois groupes.
Conclusion. Dans notre étude, les anomalies cytogénétiques de bon pronostic sont moins fréquentes que dans la littérature. La cytogénétique a un impact sur les SSE et SG. Dans notre série, il n’y a pas de différence significative en terme de SSE pour les deux groupes Int et Déf d’où l’interet de la biologie moléculaire pour mieux caractériser les groupes pronostiques.
06-34
Hétérogénéité intratumorale dans les leucémies aiguës myéloïdes : résultats d’une cohorte de vie réelle
M. Cerrano*1, M. Passet2, F. Rabian1, R. Rahmé3, E. Lengline1, E. Raffoux1, M. Sebert4, O. Maarek2, S. Mathis5, L. Ades4, P. Fenaux4, N. Boissel6, H. Dombret1, E. Clappier2, R. Itzykson1
1 Hématologie adulte, Hôpital Saint-Louis (AP-HP), Paris ; 2 Laboratoire d’hématologie, Institut Universitaire d’Hématologie (Hôpital St Louis), Paris ; 3 Hématologie seniors, Hôpital Saint-Louis, Assistance Publique Hôpitaux de Paris, Paris ; 4 Hématologie seniors, Hôpital Saint-Louis, Paris ; 5 Hématologie biologique, Hôpital Saint-Louis, Paris ; 6 Hématologie adolescents et jeunes adultes, Assistance Publique Hôpitaux de Paris, Paris
Introduction. Les LAM sont des maladies oligoclonales. L’HIT est liée à un pronostic défavorable dans de nombreux cancers. À l’exception des patients (pts) avec un caryotype complexe (Bochtler et al., 2013), les conséquences cliniques de l’HIT restent inconnues dans les LAM. À partir de données de séquençage ciblé par NGS, nous avons étudié l’HIT dans une cohorte prospective monocentrique de LAM diagnostiquées consécutivement.
Matériels et méthodes. Depuis novembre 2015, tous les pts avec un nouveau diagnostic de LAM ont été séquencés avec un panel de 60 gènes (capture Agilent SureSelect), avec une profondeur moyenne de 815×. Les fréquences d’allèle variant ont été ajustées sur le nombre de copies pour estimer pour chaque mutation la fraction de cellules leucémiques (FCL). Les mutations ont été classées par FCL décroissante puis assignées à des clones différents en cas de différence de FCL > 10 %. Les mutations ancestrales sont celles du clone primaire, les autres sont classées comme mutations secondaires. Pour chaque patient ont été dérivés le nombre total des clones et l’index HIT, défini comme le rapport nombre de mutations secondaires/nombre de mutations ancestrales (Turajlic et al., 2018).
Résultats. 201 pts (âge médian 67 ans, LAM secondaire 12,4 %) ont été inclus, avec un nombre médian de 4 mutations (range 0-12). Une hétérogénéité clonale (>1 clone) a été détectée dans 71 % des pts. 140 pts ont reçu une chimiothérapie intensive (CI), 50 des hypométhylants (HMA) et 11 un traitement de support seulement. Le risque ELN 2017 était favorable, intermédiaire, défavorable et non évaluable dans 35 %, 27 %, 35 % et 3 % des pts. Un index HIT élevé était associé à un nombre plus élevé de clones (p = 0,001) et de mutations (p < 0,001), à la présence d’au moins une mutation des gènes modificateurs d’histones (p = 0,0004), du complexe des cohésines (p = 0,0018) ou de transduction du signal (p = 0,002), mais pas aux mutations des gènes de méthylation ou du spliceosome. L’HIT était indépendante du risque ELN, de l’âge, de la leucocytose et du type de LAM (secondaire ou de novo).
Une analyse pronostique préliminaire de l’HIT a été conduite chez les 140 pts traités par CI (suivi médian 12,5 mois). 84 % ont obtenu une rémission complète (RC) et 23 % ont été allogreffés en première RC. La survie sans événements (SSE) médiane était 18,7 vs 8,0 mois chez les pts de risque ELN défavorable versus non défavorable (p = 0,016). En analyse univariée, les mutations de la méthylation (p = 0,04) et du spliceosome (p = 0,007) étaient associées à une SSE inférieure. Le nombre de mutations (p = 0,6) ou de clones (p = 0,8) n’avait pas d’impact. Les pts avec moins de mutations secondaires que de mutations ancestrales (index HIT < 1, groupe « HIT faible », 46 %) avaient une SSE médiane de 13,6 mois versus 19,8 mois pour les pts avec index HIT ≥ 1 (groupe « HIT élevée », p = 0,067). Dans un modèle multivarié incluant l’âge, la leucocytose, le risque ELN 2017, le type de LAM, la présence de mutations de méthylation ou du spliceosome, la présence d’une HIT élevée prédisait une SSE prolongée (p = 0,03), indépendamment de l’âge élevé (p = 0,07), du risque ELN défavorable (p = 0,005) et de la leucocytose (p = 0,004). La censure à l’allogreffe en première RC n’affectait pas ce résultat.
Conclusion. Nos résultats confirment que la majorité des LAM sont oligoclonales. L’HIT pourrait être associée à une survie sans événement prolongée chez les pts traités intensivement.
06-35
Leucémie aiguë au cours de la grossesse : étude rétrospective de trois cas
W. Chenbah*1, B. Achour2, H. Regaig3, N. Bensayed2, E. Bouslama2, YY. Ben2, K. Abderrahim2
1 Hématologie, Hôpital Farhat Hached Sousse, Monastir, Tunisie ; 2 Hématologie clinique, CHU Farhat Hached, Sousse, Tunisie ; 3 Hématologie, Hôpital Farhat Hached Sousse, Sousse, Tunisie
Introduction. L’association d’une leucémie aiguë (LA) et d’une grossesse est une situation très rare mettant en jeu aussi bien le pronostic maternel que fœtal. Le diagnostic est généralement fait pendant le deuxième et le troisième trimestre. Elle pose un problème éthique et thérapeutique car la chimiothérapie au cours de la grossesse expose le fœtus aux complications.
Patients et méthodes. Étude rétrospective descriptive de trois femmes enceintes atteintes de leucémie aiguë colligées au service d’hématologie à l’hôpital Farhat Hached depuis janvier 2008 à janvier 2018.
Résultats. Le diagnostic était fait au premier trimestre de la grossesse (14 SA) pour un cas et pendant le troisième trimestre pour 2 cas (27, 32 SA) chez des femmes multipares. La symptomatologie se résume à un syndrome anémique associé à un syndrome hémorragique (purpura, ecchymose) pour 2 cas et un flou visuel pour 1 cas. L’hémogramme a montré une pancytopénie associée à une blastose périphérique dans 2 cas et une hyperleucocytose à 350 000/mm3, une blastose et une anémie normocytaire dans un autre cas. Le bilan biologique en faveur d’une CIVD dans 2 cas. Le myélogramme et l’immunophénotypage étaient en faveur d’une leucémie aiguë myéloblastiques (LAM2, LAM3) dans 2 cas et leucémie aiguë lymphoblastique (LAL) dans 1 cas. Les trois patientes ont bénéficié de soins de support (transfusion de culots globulaires, plaquettaires et de PFC). La patiente présentant une LAL hyperleucocytaire a bénéficié de plusieurs séances d’échange transfusionnel, d’un traitement hypo-uricémiant pour prévenir le syndrome de lyse et d’une corticothérapie. L’accouchement est par voie basse dans 2 cas (nouveau-nés prématurés) et une interruption thérapeutique de grossesse (LAM3) en raison de l’urgence thérapeutique. Les trois patientes ont reçu une chimiothérapie après consentement éclairé après l’accouchement. L’évolution au long cours était marquée par la survenue d’un décès (LAM3) suite à des complications infectieuses lors de traitement de consolidation et d’une rémission complète pour les 2 autres patientes.
Conclusion. L’association de la leucémie aiguë et grossesse est un événement rare. Elle nécessite une prise en charge multidisciplinaire tenant compte des impératifs de la maladie et de son traitement, de la femme et de son désir de grossesse ainsi que des dimensions éthiques et morales. L’utilisation de chimiothérapie au cours de la grossesse est possible après 20 SA. L’interruption de grossesse s’impose avant cette date. Les résultats des traitements des LA pendant la grossesse se comparent favorablement à ceux obtenus dans une autre situation, à intensité égale de traitement.
06-36
Profils phénotypiques par cytométrie en flux des leucémies aiguës myéloblastiques
S. Oukid*, S. Taoussi, F. Lamraoui, N. Rekab, Y. Bouchakor Moussa, KM. Benlabiod, MT. Abad, M. Bradai
Hématologie, CAC, Laboratoire de recherche sur les hémopathies Malignes et les Hémoglobinopathies. Université, Blida I, Algérie
Introduction. L’application de la cytométrie en flux (CMF) dans les leucémies aiguës (LA) permet de mieux caractériser les cellules malignes, d’apprécier le pronostic et d’assurer une prise en charge thérapeutique plus rationnelle. L’objectif de notre étude est d’évaluer son intérêt pour le diagnostic et la classification des leucémies aiguës myéloblastiques (LAM).
Matériels et méthodes. Nous rapportons 368 cas de LAM : 30 enfants (8,2 %) : 14 filles et 16 garçons d’âge moyen de 8 ans (1-15) et 338 adultes (91,8 %) : 181 femmes et 157 hommes d’âge moyen de 46 ans (16-83). La présentation initiale est dominée par les signes d’insuffisance sanguine : 97 % et/ou un syndrome tumoral : 37 %. L’étude cytologique des frottis sanguins et médullaires a conclu à : LAM : 300 cas (81,5 %), LAL : 8 cas (2,1 %), LA indifférencié : 60 cas (16,3 %). L’immunophénotypage par CMF a été réalisé sur sang : 34 cas (9,2 %), et actuellement systématiquement sur moelle : 334 cas (90,8 %).
Résultats. Les résultats de l’immunophénotypage par CMF sont représentés dans le tableau ci-dessous.LAM M0 M1 M2 M3 M4 M5 M6 M7 M LA Enfants 1 1 9 4 9 1 2 1 1 1 Adultes 23 34 66 44 107 27 13 1 22 1 Total 24 35 75 48 116 28 15 2 23 2
Fréquence d’expression des différents marqueurs : Myéloïdes : MPO : 86 %, CD33 : 90 %, CD13 : 85 %, CD15 : 60 %, CD117 : 74 % et d’immaturité : HLA Dr : 78 % et le CD34 : 66 %. Les facteurs aberrants les plus fréquents sont : le CD7 : 21 %, CD56 : 27 %, CD4 : 31 %, CD3 : 9 %, CD2 : 7,8 %, CD10 : 4 %, CD19 : 5,7 % et CD22 : 5 %.
Discussion. Dans plus de 18 % des cas le diagnostic de LAM n’a été posé que par la CMF et dans 34 %, elle a permis de préciser le type exact de LAM sachant que la cytologie a évoqué un autre type de LAM. La CMF n’a pu préciser le type de LAM dans 25 cas (6,7). Le taux d’expression des marqueurs myéloïdes rejoint celui de la littérature. (1) La prédominance d’expression des marqueurs lymphoïdes T notée dans notre étude est retrouvée aussi dans une étude tunisienne. (1) L’expression des marqueurs d’immaturité rejoint celle de la littérature mais pas celle de la série tunisienne. (1) Dans notre série, le taux d’expression du CD56 facteur de pronostic (27 %) est nettement supérieur à la série tunisienne (15,5 %). (1).
Conclusion. La CMF, examen sensible et fiable, a été d’un apport considérable pour le diagnostic des LA ; elle complète utilement les examens cytologiques et cytochimiques. La CMF est le seul outil qui permet une étude des cas rares de LAM et plus particulièrement les LA (M0, M1, M6, M7) données indispensable à une adaptation thérapeutique et une évaluation pronostique.
06-37
Aspects clinicobiologique et évolutif des leucémies aiguës myéloïdes secondaires à un syndrome myélodysplasique : à propos de 20 cas
N. Ait Amer*, F. Tensaout, F. Belhadri, H. Moussaoui, F. Boukhemia, N. Abdennebi, S. Akhrouf, F. Harieche, RM. Hamladji, R. Ahmed Nacer
Hématologie Greffe de Moelle Osseuse, Centre Pierre et Marie Curie, Sidi M’Hamed, Algérie
Introduction. Les syndromes myélodysplasiques (SMD) sont des insuffisances médullaires qualitatives dont la complication majeure reste la transformation en leucémie aiguë (20-30 %). Le pronostic reste très sombre en raison de son caractère secondaire.
Patients et méthodes. Il s’agit d’une étude rétrospective allant de janvier 2011 à décembre 2017 portant sur une série de 20 patients (pts) présentant une leucémie aiguë myéloïde (LAM) secondaire à un SMD (multilignées : 12, AREB 1 : 3, AREB 2 : 5), après un délai moyen de 12 mois (6-36). L’âge médian des pts est de 64 ans (16-88) dont 11 pts > 65 ans et 9 pts ≤ 65 ans, le sex-ratio est de 2,3 (14 H/06 F). Le diagnostic est établi sur les données de l’hémogramme et l’analyse cytologique et cytochimique des frottis sanguin et médullaire, complétée par un immunophénotypage des blastes par cytométrie en flux. Treize pts ont reçu un traitement (TRT) spécifique (dont 2 pts ont bénéficié d’une allogreffe) : VIDAZA (n : 6), Rubidomycine 3 J-cytarabine 7 j (n : 5), cytarabine à faible dose (n : 2) ; 6 pts ont bénéficié d’un TRT symptomatique et un pt est décédé avant le TRT.
Résultats. Sur le plan clinique 12 pts (60 %) ont un ECOG ≤ 2, tous les pts ont présenté un syndrome anémique, 4 pts un syndrome infectieux, 4 pts un syndrome hémorragique et 2 pts un syndrome tumoral.
Sur le plan biologique, le taux moyen des globules blancs est de 13 G/L (3-45), une hyperleucocytose supérieure à 10 G/L chez 7 pts, le taux moyen d’hémoglobine 7 g/dL (4-10), tous les pts ont présenté une thrombopénie avec un taux moyen de plaquettes de 77 G/L (4-110). La blastose sanguine moyenne est de 20 % (2-90) et médullaire de 42 % (21-100), selon la classification FAB (LAM0 : 4, LAM1 : 1, LAM2 : 12, LAM3 : 2, LAM5 : 1).
Sur le plan évolutif, sur les 20 pts, 19 sont décédés (95 %) : 1 pt avant TRT, 6 pts sous TRT symptomatique, 3 pts en cours de TRT, 8 pts en échec, 1 pt en rémission complète (GVH aiguë) et un pt vivant en rémission complète après allogreffe de moelle osseuse.
Conclusion. L’évolution des SMD se fait de façon inéluctable vers LAM dont le pronostic reste sombre en raison de son caractère secondaire, comme en témoignent nos résultats : 95 % des pts de notre série sont décédés.
06-38
Évaluation du protocole national AML-MA 2011 de traitement de la leucémie aiguë myéloïde chez l’adulte : expérience du service d’hématologie de Casablanca
M. Dakkoune*1, M. Lamchahab1, BK. Ait2, MR. Massi1, S. Cherkaoui1, N. Khoubila1, Q. Meryem1, M. Rachid1, A. Madani1, M. Harif1, Q. Asmaa1
1 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc ; 2 Laboratoire de génétique et pathologie moléculaire, CHU Ibn Rochd Casa, Casablanca, Maroc
Introduction. La leucémie aiguë myéloïde (LAM) représente 1 % des cancers et 80 % des leucémies aiguës de l’adulte dont l’incidence est en constante augmentation. Le traitement des LAM dans les pays en développement reste un challenge. L’objectif du protocole AML MA-2011 national a été d’obtenir plus de 70 % de rémission complète, moins de 10 % de décès au cours des inductions, et l’amélioration du taux de survie sans événement à 4 ans à 40 %.
Le but de notre travail était d’évaluer les résultats thérapeutiques du protocole national AML-MA 2011 chez l’adulte au sein de service d’hématologie-oncologie pédiatrique de l’hôpital de 20-Août de Casablanca.
Patients et méthodes. Il s’agit d’une étude descriptive rétrospective, s’étalant sur une période de 6 ans, de janvier 2011 au décembre 2016 au sein de service d’hématologie et d’oncologie pédiatrique (SHOP).
Les critères d’inclusions :
– âge entre 18 ans et 60 ans.
– LAM de novo.
– absence de traitement antérieur pour la LAM.
– traitement selon le protocole AML-MA 2011.
– la rémission complète (RC) est définie par la présence de moins de 5 % de myéloblastes dans la moelle osseuse.
Les données épidémiologiques, cliniques et thérapeutiques des patients atteints de LAM ont été collectées à l’aide d’une fiche d’exploitation à partir des dossiers médicaux des malades. L’analyse statistique a été effectuée par SPSS 18,0. Les courbes de survie ont été réalisées par la méthode non paramétrique de Kaplan-Meier.
Résultats. 760 patients adultes ont été diagnostiqués LAM durant la période d’étude, 383 ont été inclus dans notre étude. L’âge médian était de 39 ans (18-60), le sex-ratio H/F était de 1,04. La leucocytose moyenne était de 37,4 G/L. Selon la classification FAB, les types cytologiques dominants étaient M2 : 126 (33 %), M1 : 118 (31 %), M4 : 55 (14,4 %). L’immunophénotypage était réalisé chez 307 (80,1 %) patients et le caryotype était fait et réussi chez 361 malades, 60 patients (16,7 %) étaient de groupe pronostic favorable, 224 (62 %) de groupe intermédiaire, 77 (21,3 %) de groupe défavorable. La rémission complète (RC) post induction I, était obtenue chez 197 malades (51,4 %), 121 (31,6 %) patients sont décédés au cours des inductions (I et II), dont 42 % étaient par infection. La RC était maintenue chez 122 (31,9 %) des patients avec un recul médian de 3 ans, 39 (10,2 %) des patients avaient rechuté avec un recul médian de 1,23 an. La survie sans événement à 3 ans était estimée à 40 % pour le groupe favorable, 22,3 % pour le groupe intermédiaire et 18,2 % pour le groupe défavorable.
Conclusion. Les résultats du traitement des adultes atteints de LAM par le protocole AML-MA-2011 reste insuffisantes, d’où l’intérêt de l’adaptation du traitement aux facteurs pronostiques en particulier le profil cytogénétique et moléculaire, réduire la toxicité de la chimiothérapie par une amélioration des soins de support avec la prévention et la gestion des infections associées aux soins.
06-39
Apport de la RT-MLPA dans la prise en charge des leucémies aiguës lymphoblastiques Philadelphie-like de l’adulte
FX. Gros*1, C. Moreau2, T. Leguay1, E. Klein2, A. Bidet2
1 Hématologie, CHU de Bordeaux, Bordeaux ; 2 Laboratoire d’hématologie, CHU de Bordeaux, Bordeaux
Introduction. La leucémie aiguë lymphoblastique Philadelphie-like (LAL Ph-like) est une entité provisoire dans la classification OMS 2016, présentant différentes altérations biologiques à l’origine d’une expression génique se rapprochant des LAL à chromosome de Philadelphie (LAL Ph+) et caractérisée par de nombreuses altérations au niveau des voies ABL et JAK/STAT constituées en majorité de gènes de fusion avec de multiples partenaires. La technique de RT-MLPA décrite initialement par Ruminy et al. en 2016 permet d’identifier de manière rapide et fiable un très grand nombre de transcrits chimériques impliqués notamment dans ces LAL Ph-like1. Cette étude a pour objectif de démontrer la faisabilité d’un dépistage des LAL Ph-like par RT-MLPA associée à la FISH IGH pour ne pas méconnaître les remaniements IGH-CRLF2 et IGH-EPOR notamment, qui ne sont pas des transcrits, dans la LAL de l’adulte et l’impact pronostique de cette recherche sur une cohorte monocentrique.
Matériels et méthodes. De 2008 à 2018, parmi les patients de plus de 25 ans pris en charge pour une LAL-B dans notre centre, du matériel génétique était disponible pour analyse pour 80 patients dont 44 présentaient une anomalie génétique récurrente et ont donc été exclus de la recherche d’anomalies Ph-like. Les 36 patients sans anomalies récurrentes, appelés « B-other » selon la classification OMS 2016, ont bénéficié rétrospectivement de la technique RT-MLPA suivie d’une FISH IGH en cas de négativité.
Résultats. Sur les 36 patients testés, 8 (22 %) présentaient une anomalie du spectre des anomalies Ph-like (3 réarrangements P2RY8-CRLF2, 3 réarrangements IGH-CRLF2, une fusion BCR-FGFR1 et une fusion EBF1-PDGFRB). L’âge médian des patients Ph-like était de 63,5 ans [IIQ 58-72] versus 52 ans [IIQ 39-63] pour les patients non Ph-like. Avec un suivi médian de 4,7 ans, la survie sans événement médiane des patients Ph-like était significativement inférieure à celle des autres patients « B-Other » (1 an vs non atteinte, p = 0,017).
Discussion. Il existe très peu de données dans la littérature sur le profil des patients adultes avec LAL Ph-like par rapport aux patients pédiatriques. Notre étude retrouve une proportion importante de patients Ph-like (22 %) parmi les patients identifiés comme « B-Other », soit 10 % de l’ensemble des LAL-B adultes au diagnostic dans notre centre sur les 10 dernières années. Ces patients présentent un âge plus élevé, et, comme les patients pédiatriques une présentation plus volontiers hyperleucocytaire avec une moins grande sensibilité à la chimiothérapie et une survie diminuée. Il n’existe actuellement pas de consensus sur le périmètre exact et les techniques de détection des anomalies Ph-like. L’étude de transcription génique, considérée comme le standard, est longue, coûteuse et non utilisée en routine en France. Notre technique permet de détecter une proportion importante de patients avec un profil de LAL Ph-like tout en permettant une utilisation en routine avec un coût maîtrisé. Cette détection, outre l’intérêt pronostique, pourrait permettre à cette population de bénéficier de thérapies ciblées selon les anomalies détectées.
Conclusion. Le dépistage des LAL Ph-like de l’adulte par RT-MLPA associée à la FISH est faisable en routine et permet de discriminer une population Ph-like à haut risque de rechute, majoritairement d’âge > 60 ans. Rarement accessibles à une chimiothérapie intensive toxique, ces patients pourraient bénéficier de thérapies ciblées sur les anomalies dépistées.
06-40
Association blinatumomab/ponatinib en traitement de rattrapage des leucémies aiguës lymphoblastiques B à chromosome Philadelphie positif réfractaires ou en rechute
MA. Couturier*1, X. Thomas2, F. Huguet3, C. Berthon4, C. Simand5, Y. Hicheri6, M. Hunault-Berger7, P. Chevallier8
1 Hématologie clinique, CHRU de Brest, hôpital Morvan, Brest ; 2 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 3 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 4 Maladie du sang, CH Régional Universitaire de Lille, Lille ; 5 Département d’oncohématologie, Hôpitaux Universitaires de Strasbourg, Strasbourg ; 6 Hématologie, CHU de Montpellier, Montpellier ; 7 Service des maladies du sang, CHU - CHU Angers, Angers ; 8 Service d’hématologie clinique, Hôtel-Dieu, Nantes
Introduction. Malgré les progrès obtenus par l’association d’inhibiteurs de tyrosine kinase anti-BCR-ABL1 (ITK) à une polychimiothérapie, le pronostic des LAL Ph+ réfractaires ou en rechute reste très péjoratif, avec une médiane de survie de 3 à 7 mois (1, 2). Toutefois, l’arsenal thérapeutique s’est récemment enrichi : le ponatinib, un ITK de troisième génération actif notamment sur les mutations T315I, a ainsi fait la preuve de son efficacité (2), et des résultats encourageants ont été rapportés avec le blinatumomab (anticorps monoclonal bispécifique anti-CD3/CD19) en monothérapie (3). L’objectif de notre étude était d’évaluer l’efficacité et la tolérance d’une association blinatumomab + ponatinib (blina/pona).
Patients et méthodes. Identifiés rétrospectivement dans 8 centres français, 15 patients ont reçu l’administration concomitante de blinatumomab en injection continue à 28 mg/jour, 28 jours/6 semaines, et de ponatinib en prise orale quotidienne, pour une LAL Ph+ réfractaire ou en rechute.
Résultats. Neuf hommes et 6 femmes ont été analysés, avec un âge médian au diagnostic de 53 ans [17 ; 72] : 2 patients présentaient une LMC en phase blastique et 13 une LAL Ph+ de novo, 5 avaient une mutation BCR-ABL1 T315I. L’association blina/pona a été administrée après une première (n = 6) ou une deuxième rechute (n = 8), un patient était réfractaire. Respectivement 9 et 3 patients avaient déjà reçu une allogreffe ou une autogreffe de cellules-souches hématopoïétiques (CSH), la majorité (n = 12) avaient reçu 2 lignes d’ITK ou plus. Trois cycles de blinatumomab ont été délivrés [1 ; 6] alors que le ponatinib a été administré en continu à la dose de 45 mg/j chez 10 patients (67 %) et 30 mg/j chez 5 patients (33 %), pendant une durée médiane de 4 mois [1 ; 17].
La tolérance a été satisfaisante : tous les patients ont pu recevoir un premier cycle complet sans effet indésirable de grade 3 ou 4. Après le cycle n̊1, le blinatumomab a été interrompu dans 47 % des cas du fait d’effets indésirables neurologiques (n = 4) ou infectieux (n = 3) ; le ponatinib a été arrêté dans 33 % des cas du fait d’effets indésirables neurologiques (n = 3), de rétention hydrosodée (n = 1) et d’une artériopathie sévère. Tous les événements neurologiques ont totalement régressé après interruption du blinatumomab ou du ponatinib.
Tous les patients (sauf un) ont obtenu une rémission complète (RC) cytologique (93 %) ; parmi eux, 12/14 (86 %) ont obtenu une RC moléculaire (RCM). Cependant, 2 patients présentaient une rechute neuroméningée, traitée par chimiothérapie intrathécale. Six patients ont pu recevoir une allogreffe de CSH. À partir du premier cycle de blina/pona, la survie globale était de 18,2 mois [8,5 ; non atteinte] et la survie sans leucémie de 14,7 mois [4,9 ; non atteinte]. À la date des dernières nouvelles (novembre 2018), 6 rechutes sont survenues 7 mois [2 ; 15] après le début du traitement par blina/pona, 1 patient restait non-répondeur. Six patients sont décédés (40 %) de complications infectieuses bactériennes (n = 3) ou fongiques (n = 1), d’un second cancer (n = 1) ou de l’évolution de la LAL (n = 1). Parmi les 9 patients vivants, un présente une rechute cytologique et 8 sont toujours en RCM. Cependant, 2 ont une rechute neuroméningée isolée. Quatre patients poursuivent actuellement un traitement de maintenance par ponatinib en monothérapie.
Conclusion. L’association blina/pona semble efficace et bien tolérée dans le traitement des LAL Ph+ réfractaires ou en rechute, et pourrait être proposée en première ligne thérapeutique. Des études prospectives sur de plus larges cohortes sont nécessaires pour confirmer nos résultats.
06-41
ATG2B/GSKIP dans les leucémies aiguës myéloïdes de novo : prévalence élevée de la prédisposition génétique aux Antilles et rôle potentiel de la surexpression dans les leucémies aiguës myéloïdes acquises
J. Pegliasco1, B. Schmaltz-Panneau2, JE. Martin1, S. Chraibi1, C. Legoupil3, P. Helias4, F. Pasquier1, V. Saada5, JH. Bourhis1, N. Auger6, J. Delaunay7, P. Cornillet-Lefebvre8, C. Récher9, W. Vainchenker10, S. De Botton11, C. Marzac5, JC. Méniane12, I. Plo2, C. Bellanné-Chantelot13, JB. Micol*1
1 Service d’hématologie clinique, Gustave Roussy, Villejuif ; 2 Inserm, UMR1170, Université Paris-Sud, Gustave Roussy, Villejuif ; 3 Service de biostatistiques, Gustave Roussy, Villejuif ; 4 Cancérologie, CHU de la Guadeloupe, Pointe-à-Pitre ; 5 Hématologie biologique, Gustave Roussy, Villejuif ; 6 Département de biologie-pathologie, Gustave Roussy, Villejuif ; 7 Service d’hématologie clinique, Hôtel-Dieu, Nantes ; 8 Biologie, Hôpital Robert Debré, Reims ; 9 Hématologie adulte, Institut Universitaire du Cancer de Toulouse-Oncopole, Toulouse ; 10 Inserm U1170, Gustave Roussy, Villejuif ; 11 Service d’hématologie clinique, Institut Gustave Roussy, Villejuif ; 12 Pôle de cancérologie hématologie urologie, CHU de Fort de France, Fort-de-France, Martinique ; 13 Département de Génétique, AP-HP, Hôpital Pitié-Salpêtrière, Département de Génétique, Paris
Introduction. Une duplication germinale d’une région de 700 kb du chromosome 14 (dup14) incluant les gènes ATG2B/GSKIP a été récemment décrite (Saliba et al., Nat Gen 2015) chez des patients antillais atteints de néoplasmes myéloprolifératifs (NMP) familiaux d’évolution fréquente en LAM. Nous avons ici, recherché la présence de cette dup14 chez des patients antillais atteints d’hémopathies malignes aiguës (HMA) puis étudié, dans une cohorte de LAM de novo le niveau d’expression d’ATG2B/GSKIP.
Patients et méthodes. Il s’agit d’une étude rétrospective de 100 patients adultes antillais (Martinique n = 58, Guadeloupe n = 28, autres n = 14) atteints de LAM (n = 78), leucémie aiguë lymphoblastique (n = 20), lymphome lymphoblastique (n = 3) ou leucémie aiguë de lignée ambiguë (n = 2), traités à Gustave Roussy (Villejuif) et dans les CHU de Martinique et Guadeloupe de mai 2000 à mai 2018. Les patients avec antécédent de NMP ou de myélodysplasie ont été exclus. La présence de la dup14 a été recherchée par PCR sur ADN leucocytaire.
Dans un deuxième temps, nous avons étudié, au sein d’une autre cohorte, l’expression d’ATG2B/GSKIP dans 46 LAM de novo à caryotype normal ou trisomie 8 (échantillons de la Goelamsthèque) par RT-PCR quantitative.
Résultats. Nous avons détecté par PCR six patients portant la dup14. Tous étaient atteints de LAM (aucune avec cytogénétique favorable) et originaires de Martinique. Ils représentaient 14 % des 43 LAM de Martinique, 2 n’avaient pas d’antécédent familial d’HM et 2 nouvelles familles ont été découvertes. Le caryotype était normal pour 4 patients, avec une monosomie 7 pour 2 patients (comparativement les 72 autres LAM non dup14 étaient favorables (15 %), intermédiaire (60 %) et défavorable (25 %)). Le séquençage par NGS montrait un profil somatique distinct du reste de la cohorte. Tous présentaient des mutations dans des gènes codant des facteurs d’épigénétiques et/ou de l’épissage (IDH n = 3, TET2 n = 3, ASLX1 n = 3, SRSF2 n = 3) mais aucune mutation de JAK2, MPL, CALR, TP53, RUNX1, DNMT3A et FLT3-ITD. Cinq des 6 pts ont reçu un traitement intensif, 4 ont atteint la rémission complète, 2 ont été allogreffés contre 82 %, 78 % et 29 % des LAM non dup14. Les patients avec la dup14 avaient une médiane de survie globale (SG) et de survie sans progression (SSP) de 19 (6,5-29) et 11,4 mois (6,5-29) respectivement, comparé à 52,6 (22,5-52,5) et 30 mois (15,6-60) pour ceux sans dup14.
Nous avons ensuite évalué l’expression d’ATG2B/GSKIP dans une cohorte de 46 LAM de novo (protocole GOELAMS-LAM-IR-2006). Aucune dup14 n’a été détectée. Le niveau médian d’expression d’ATG2B et GSKIP était de 23 (0,01-280) et 5,3 (0,01-50), respectivement, et corrélé entre les 2 gènes (Pearson à 0,55, régression linéaire p < 0,001). L’expression d’ATG2B/GSKIP était corrélée avec la leucocytose (p = 0,003 et p = 0,07). Nous n’avons pas trouvé d’effet sur la SG ni la SSP.
Conclusion. Nous décrivons, pour la première fois, l’existence dans une proportion importante d’une prédisposition génétique incluant ATG2B/GSKIP dans une population de patients Martiniquais (14 %). De plus, l’étude de l’expression d’ATG2B/GSKIP suggère que leur rôle dans la leucémogenèse ne se limiterait pas aux patients porteurs de la dup14 germinale.
06-42
La mutation PAX5P80R définit un nouveau sous-type de leucémie aiguë lymphoblastique B de pronostic favorable chez l’adulte : une étude du GRAALL
M. Passet*1, N. Boissel2, F. Sigaux3, C. Saillard4, I. Ba1, X. Thomas5, Y. Chalandon6, T. Leguay7, E. Lengline2, J. Konopacki8, S. Quentin1, E. Delabesse9, M. Lafage-Pochitaloff10, C. Pastoret11, N. Grardel12, V. Asnafi13, V. Lheritier14, J. Soulier1, H. Dombret2, E. Clappier1, au nom du Group for Research on Adult Acute Lymphoblastic Leukemia (Graall)
1 Laboratoire d’hématologie, Inserm U944, Hôpital Saint-Louis (AP-HP), Université Paris Diderot, Institut Universitaire d’Hématologie, Paris ; 2 Département d’hématologie, EA-3518, Hôpital Saint-Louis (AP-HP), Université Paris Diderot, Institut Universitaire d’Hématologie, Paris ; 3 Université, Paris Diderot, Commissariat à l’Énergie Atomique et aux énergies renouvelables, Paris, Saclay ; 4 Hématologie 2, Institut Paoli Calmettes, Marseille ; 5 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 6 Département d’hématologie, Hôpital et Université de Genève, Genève, Suisse ; 7 Hématologie, CHU de Bordeaux, Bordeaux ; 8 Hématologie, Hôpital d’Instruction des Armées Percy, Clamart ; 9 Oncopole, CHU Toulouse - Casselardit Ancely, Toulouse ; 10 Laboratoire de cytogénétique oncohématologique, Hôpital de la Timone, Marseille ; 11 Laboratoire d’Hématologie, CHU de Rennes, Rennes ; 12 Laboratoire d’hématologie, CH Régional Universitaire de Lille, Lille ; 13 Hématologie, Hôpital Necker-Enfants Malades, AP-HP, Paris ; 14 Coordination du groupe Graall, Centre Hospitalier Lyon Sud, Lyon
Introduction. Les leucémies aiguës lymphoblastiques de la lignée B (LAL-B) sont caractérisées par des anomalies chromosomiques récurrentes qui définissent des groupes homogènes d’un point de vue biologique mais aussi clinique et pronostique (anomalies classantes). Leur identification est donc importante pour optimiser la prise en charge thérapeutique. Cependant, environ la moitié des LAL-B de l’adulte ne présente aucune des anomalies classantes identifiées à ce jour par les analyses cytogénétiques et moléculaires standard. Afin de caractériser ces cas nous avons effectué une étude globale basée sur le séquençage nouvelle génération du transcriptome d’une grande série de LAL-B.
Patients et méthodes. Une cohorte exploratoire de 170 LAL-B sans anomalie classante a été étudiée par RNA-seq (librairie TruSeq, séquençage Illumina) avec analyses transcriptionnelle et mutationnelle. Une cohorte additionnelle de 419 patients inclus dans les protocoles GRAALL-2003, -2005 et -2014 a été criblée par séquençage Sanger pour la mutation PAX5 P80R. Les anomalies génomiques associées ont été recherchées par séquençage ciblé de 160 gènes (librairie SureSelect Agilent, séquençage Illumina) et CGH-array (puce pangénomique 400K, Agilent). Les caractéristiques cliniques et le pronostic ont été évalués chez 312 patients inclus dans les protocoles GRAALL-2003 et -2005.
Résultats. L’analyse du transcriptome par clustering non supervisé a permis d’identifier un nouveau groupe, en plus de ceux décrits récemment (BCR-ABL1-like, DUX4/ERG et ZNF384). Une mutation unique de PAX5, c.239C>G, p.P80R, a été mise en évidence dans chacun des 14 cas de ce nouveau groupe. PAX5 est un facteur de transcription essentiel à la différenciation lymphoïde B et altéré de façon récurrente dans les LAL-B par délétions génomiques ou mutations perte de fonction. La modélisation de la mutation PAX5 P80R et la signature transcriptomique associée suggèrent une perte de fonction partielle de PAX5 mais également l’activation d’un programme transcriptionnel aberrant. La mutation PAX5 P80R a été retrouvée chez 16 patients de la cohorte additionnelle, jamais en association avec une anomalie classante, confirmant que cette mutation définit un groupe distinct de LAL-B qui représente 5,3 % (29/551) des LAL-B Ph-négatives. La mutation PAX5 P80R est associée à un spectre très caractéristique d’altérations additionnelles : inactivation du deuxième allèle de PAX5, mutations des facteurs de signalisation Ras ou CRLF2/IL7R-JAK-STAT, délétions de CDKN2A souvent en rapport avec des anomalies 9p. Des délétions complètes d’IKZF1 par del(7p) sont fréquemment observées, contrairement aux délétions intragéniques (0 % vs 27,0 %, P = 0,025). Les caractéristiques clinicobiologiques au diagnostic du groupe PAX5 P80R n’étaient pas différentes du reste de la cohorte. En revanche, une meilleure réponse moléculaire post-induction (MRD1 ≥ 10-3, 0 % vs 31,2 %, P = 0,036), une meilleure survie sans événement (EFS à 5 ans, 81,8 % vs 42,7 % ; HR = 0,20, 95% CI 0,05-0,83 ; P = 0,026) et une meilleure survie globale (OS à 5 ans 91 % versus 47 %, HR = 0,12, 95% CI 0,02-0,87 ; P = 0,036) ont été observées pour les patients PAX5 P80R traités dans les protocoles GRAALL-2003/2005.
Conclusion. La mutation PAX5 P80R caractérise un nouveau sous-type oncogénétique de LAL-B. Il s’agit du premier sous-type de LAL-B défini par une mutation ponctuelle, qui est également le premier sous-type de pronostic favorable chez l’adulte.
06-43
Aspects immunophénotypiques par cytométrie en flux des leucémies aiguës biphénotypiques
S. Oukid*, S. Taoussi, F. Lamraoui, N. Rekab, Y. Bouchakor Moussa, KM. Benlabiod, MT. Abad, M. Bradai
Hématologie, CAC, Laboratoire de recherche sur les hémopathies Malignes et les Hémoglobinopathies. Université, Blida I, Algérie
Introduction. L’immunophénotypage par cytométrie en flux (CMF), outil essentiel pour le diagnostic des leucémies aiguës (LA), a individualisé une nouvelle entité : la leucémie aiguë biphénotypique (BAL) ou mixte. Notre étude présente les caractéristiques de cette entité que nous avons relevées dans notre cohorte de patients.
Matériels et méthodes. Notre étude a porté sur 41 patients atteints de BAL : 5 enfants : 3 garçons âgés de 3 jours, 5 et 12 ans et 2 filles âgées de 4 et 5 ans et de 36 adultes : 13 femmes et 23 hommes d’âge moyen de 41 ans (16-64). La présentation initiale est dominée par les signes insuffisance sanguine dans 35 cas et/ou un syndrome tumoral dans 25 cas. Une hyperleucocytose dans 31 cas, une thrombopénie dans 34 cas et une anémie dans 33 cas. Une blastose médullaire moyenne est de l’ordre de 82 %. La cytologie a révélé une LAM dans 16 cas, une LAL ans 10 cas et une LA indifférenciée dans 15 cas ; la myéloperoxydase était positive dans 35 % des cas. Les prélèvements pour CMF sont faits sur sang périphérique : 8 cas (19,5 %) et sur prélèvement médullaire : 33 cas (80,5 %) avec une richesse médullaire moyenne de 157 594 elts/μl. Les étapes suivantes sont : filtration de la moelle, numération, dilution, marquage par un panel large d’anticorps monoclonaux pour cibler les populations Lymphoïdes (B, T, NK) et Myéloïdes, acquisition et analyse sur un cytométre en flux.
Résultats. La CMF montre 2 groupes phénotypiques qui sont illustrés dans les tableaux (1-2) :
premier groupe : LAM et LAL T : 26 cas (63,5 %). deuxième groupe : LAM et LAL B : 15 cas (36,5 %). Score d’EGIL des LAM et LAL B et TScore EGIL LAM/LAL T LAM/LAL B Moyenne d’expression Myéloïdes 3,2 Myéloïdes 3,7 Lymphoïdes T 4,8 Lymphoïdes B 4,7 Score ≥ 4 Myéloïdes 2 cas Myéloïdes 8 cas Lymphoïdes T 8 cas Lymphoïdes B 12 cas Tableau 1
Discussion. Nos résultats rejoignent la littérature en termes de données épidémiologiques (fréquence, âge, sexe). Comparativement aux résultats maghrébins on note sur le plan phénotypique une fréquence plus élevée des BAL avec coexpression des marqueurs myéloïdes et Lymphoïdes T dans notre série, ceci s’expliquerait en partie par la fréquence plus élevée des leucémies aiguës lymphoblastiques T dans notre région (49 %) (4) vs (20 %) en France (3). Notre série est caractérisée par la grande difficulté du classement morphologique, dans prés de 36 % des cas par opposition aux séries tunisienne (2) et marocaine (1) où le type cytologique n’a pas pu être donné dans respectivement 26 % et 17 % des cas.
Conclusion. La CMF est actuellement le seul outil performant pour individualiser les leucémies aiguës.
Biphénotypiques. Le profil immunophénotypique de ces entités est important à déterminer pour la conduite thérapeutique et le pronostic.
06-44
Impact pronostique de la signature cellule souche leucémique (LSC17) dans les leucémies aiguës myéloïdes du sujet âgé de 18 à 60 ans (étude ALFA-0702)
N. Duployez*1, X. Thomas2, M. Figeac3, S. De Botton4, C. Villenet3, L. Fenwarth1, J. Lambert5, T. Braun6, N. Cambier7, I. Plantier8, F. Nguyen-Khac9, L. Gastaud10, JV. Malfuson11, T. Cluzeau12, O. Hermine13, N. Boissel14, B. Quesnel15, M. Cheok16, H. Dombret14, C. Preudhomme1
1 Laboratoire d’Hématologie, CHU Lille, Lille ; 2 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 3 Plateforme de génomique, IRCL, Institut pour la Recherche sur le Cancer de Lille, Lille ; 4 Service d’hématologie clinique, Institut Gustave Roussy, Villejuif ; 5 Hématologie, CH de Versailles, Versailles ; 6 Hématologie, Hôpital Avicenne, Bobigny ; 7 Service d’hématologie clinique, CH De Valenciennes, Valenciennes ; 8 Service d’hématologie clinique, CH de Roubaix, Roubaix ; 9 Service d’hématologie biologique, AP-HP La Pitié-Salpêtrière, Paris ; 10 Oncologie, Centre Antoine Lacassagne, Nice ; 11 Hématologie, Hôpital d’Instruction des Armées Percy, Clamart ; 12 INSERM U1065, Centre Méditerranéen de Médecine Moléculaire, Nice ; 13 Hématologie, Hôpital Necker, Paris ; 14 Département d’hématologie, Hôpital Saint-Louis, AP-HP, Paris ; 15 Service des maladies du sang, CH Régional Universitaire de Lille, Lille ; 16 ULR-S 1172, Inserm, Lille
Introduction. Les leucémies aiguës myéloïdes (LAM) sont un groupe hétérogène d’hémopathies malignes, supporté par la grande diversité des anomalies génétiques sous-jacentes à la base de la stratification thérapeutique. En dépit des progrès, elles restent des affections de mauvais pronostic, les échecs d’induction et les rechutes après obtention d’une rémission complète constituant les principaux obstacles à la guérison, y compris chez des patients sans facteur de risque défavorable. Dans ce contexte, l’identification de nouveaux biomarqueurs préthérapeutiques est essentielle pour envisager rapidement une alternative chez les patients à haut risque, essentiellement dans le cadre d’essais cliniques.
Patients et méthodes. Au total, 461 prélèvements au diagnostic de LAM étaient disponibles pour cette étude parmi les 713 patients inclus dans le protocole ALFA-0702. La signature LSC17 a été évaluée par la mesure de l’expression génique ciblée sur 17 gènes par technologie de multiplexage NanoString®. Les 2 groupes de scores LSC17 hauts et bas ont été définis de part et d’autre de la médiane comme précédemment décrit (Ng et al. Nature 2016).
Résultats. L’augmentation du score LSC17 apparaît corrélée à l’augmentation du risque ELN (figure 1A). Dans la cohorte totale (n = 461), un score LSC17 haut est associé à un pronostic plus péjoratif en survie sans maladie (HR = 1,713 [95%CI = 1,27-2,311]; p = 0,0036 ; figure 1B) et en survie globale (HR = 1,797 [95%CI = 1,341-2,409]; p < 0,0001 ; figure 1C). Ceci reste valable lorsque l’on considère uniquement les LAM à caryotype normal (n = 256) en survie sans maladie (HR = 2,01 [95%CI = 1,32-3,05]; p = 0,0036) et en survie globale (HR = 2,14 [95%CI = 1,40-3,27]; P = 0,0003). Ces résultats ne sont pas modifiés lorsque l’analyse est effectuée en censurant les patients au moment de la greffe de CSH.
Conclusion. La mesure de la signature LSC17 constitue un paramètre pronostique dans les LAM de l’adulte. La technologie NanoString® permet aujourd’hui d’accéder rapidement à cette signature transcriptomique au diagnostic de LAM, permettant d’entrevoir une alternative thérapeutique rapide et ainsi limiter les risques d’échec ou de rechute, de traitement plus difficile. Cette étude rétrospective d’une large cohorte de patients adultes traités dans le cadre d’un essai thérapeutique national pose les bases pour une évaluation prospective et l’intégration de la transcriptomique dans la future prise en charge des patients atteints de LAM.

06-45
PAX5-ELN induit une leucémie aiguë lymphoblastique de type B chez la souris dans un processus multi-étapes
L. Jamrog1, G. Chemin2, V. Fregona1, S. Hébrard1, N. Prade3, S. Lagarde3, C. Oudinet2, M. Pasquet4, S. Mancini5, E. Delabesse3, AA. Khamlichi2, B. Gerby1, C. Broccardo*1
1 Équipe : altération des facteurs de transcription dans les leucémies aiguës, Centre de Recherches en Cancérologie de Toulouse - CRCT, Toulouse ; 2 Équipe : régulation de l’instabilité génétique et de la transcription, CNRS - IPBS, Toulouse ; 3 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 4 Service d’hématologie et immunologie pédiatrique, Hôpital pour enfants, Toulouse ; 5 Équipe : interactions leuco/stromales dans l’hématopoïèse normale et pathologique, CRCM-Inserm U1068 ; Institut Paoli-Calmettes ; Aix-Marseille Université UM 105 ; CNRS UMR7258, Marseille
Introduction. PAX5 est un facteur de transcription crucial pour le maintien de l’identité des cellules lymphoïdes B. Il est la cible la plus fréquente des mutations somatiques dans les leucémies aiguës lymphoblastiques de type B (LAL-B), étant altéré dans environ 30 % des cas pédiatriques et adultes. Parmi ces altérations, diverses translocations chromosomiques résultent en la fusion d’une partie de PAX5 avec des partenaires de nature variée. Ces réarrangements chromosomiques représentent 2-3 % des LAL-B et sont considérés comme des événements oncogéniques primaires sur des bases d’études cytogénétiques. La fonction des protéines de fusion impliquant PAX5 dans l’initiation et la transformation des LAL-B reste largement méconnue. Nous avions montré que la translocation chromosomique récurrente t(7;9)(11;P13) présente chez 2 patients atteints de LAL-B fusionnait une partie de PAX5 avec la séquence codant l’élastine (ELN).
Ce projet visait à étudier l’impact de la protéine de fusion PAX5-ELN dans le développement de cellules B murines, à décrypter son rôle moléculaire et à déterminer si l’expression de PAX5-ELN est un événement princeps dans le processus leucémique.
Matériels et méthodes. Nous avons engendré un modèle de souris knock-in dans lequel l’ADNc humain de PAX5-ELN a été inséré, par recombinaison homologue, dans le locus de la chaîne lourde des immunoglobulines (IgH), en aval de l’enhancer Eμ, ce qui assure une expression restreinte aux cellules B et précoce dans la différenciation B rappelant le profil d’expression physiologique de PAX5.
Résultats. Les souris exprimant PAX5-ELN développent des LAL-B avec une incidence supérieure à 80 % et une médiane de survie de 170 jours. La transformation leucémique se produit systématiquement avec une latence minimale de 3 mois définissant une phase préleucémique suggérant l’exigence d’événements oncogéniques additionnels. Le séquençage de l’exome des souris malades a révélé que la leucémie induite par PAX5-ELN est associée à des mutations secondaires récurrentes sur les gènes Ptpn11, Kras, Pax5 et Jak3 affectant les voies de signalisation clés nécessaires à la prolifération cellulaire. De façon intéressante, l’étude génomique d’une cohorte de 101 patients pédiatriques LAL-B a montré que ce sont les mêmes altérations oncogéniques récurrentes qui y sont retrouvées.
Par des études fonctionnelles ex vivo et in vivo, nous avons démontré que PAX5-ELN affecte le développement lymphoïde B avec un blocage partiel de la différenciation B et une expansion aberrante du compartiment pro-B à l’équilibre et ce, dès le stade préleucémique. Ces pro-B ont une capacité d’autorenouvellement aberrante après transplantation. Une analyse du transcriptome des cellules pro-B préleucémiques a montré que PAX5-ELN n’agit pas comme compétiteur de PAX5 sauvage mais induit son propre programme génique.
Conclusion. Les souris exprimant PAX5-ELN représentent un modèle intéressant de LAL-B reproduisant les étapes successives de la pathologie humaine. La phase préleucémique permet d’identifier la cellule d’origine de la leucémie et la phase leucémique permet de déterminer les relais moléculaires oncogéniques ou les événements coopératifs de PAX5-ELN, potentielles cibles thérapeutiques (Jamrog et al., 2018).
06-46
Devenir des patients suivis pour leucémie aiguë myéloïde avec caryotype normal
I. Ben Amor*1, I. Frikha1, M. Ghorbel1, Z. Mlayah1, H. Bellaaj1, H. Sennana2, N. Benabdejlil3, A. Saad2, T. Ben Othman4, M. Elloumi5
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Cytogénétique, génétique moléculaire et biologie de la reproduction, CHU Farhat Hached, Sousse, Tunisie ; 3 Centre de greffe de moelle osseuse, Centre de greffe de moelle osseuse, Tunis, Tunisie ; 4 Centre national de greffe de moelle osseuse, CENTRE NATIONAL DE GREFFE DE LA MOELLE OSSEUSE TUNIS, Tunis, Tunisie ; 5 Service d’hématologie, CHU Hédi Chaker, Sfax, Tunisie
Introduction. La leucémie aiguë myéloïde (LAM) avec un caryotype normal constitue environ 40 % de toutes les leucémies aiguës myéloïdes, il s’agit d’un groupe hétérogène dont le traitement était longtemps standardisé jusqu’à l’avènement de la stratification moléculaire.
L’objectif de ce travail est de rapporter les résultats thérapeutiques des patients traités au service hématologie de Sfax pour LAM avec caryotype normal.
Matériels et méthodes. Notre étude est rétrospective, elle a concerné les adultes jeunes âgés entre 20 et 55 ans traités pour une LAM avec un caryotype normal au service hématologie de Sfax entre 2004 et 2016. Tous les patients ont reçu une cure d’induction associant aracytine et idarubicine (3+7). Le traitement postinduction comportait 3 cures de consolidations inspirées du protocole MRC (ADE, MACE, MIDAC). Une allogreffe de moelle osseuse était proposée pour les patients < 45 ans disposant d’un donneur HLA compatible.
Résultats. Nous avons recensé 62 cas des LAM avec caryotype normal parmi 142 cas de LAM entre 2005 et 2016 soit une fréquence de 43 %. L âge médian était de 37 ans avec un sex-ratio de 1,3. Le type cytologique le plus fréquent était LAM2 (23 %). Le taux de rémission complète était de 71 %. Le taux de décès en induction était de 9 %. Deux patients sont décédés au cours des cures de consolidation. L’allogreffe a été réalisée chez 15 patients soit 40 % des patients en indication d’AGMO. Un patient allogreffé était décédé en rémission. Le taux global de rechute était de 36 %, il était de 52 % dans le bras des patients non greffés alors qu’il n’était que de 8 % dans le bras des patients greffés avec p = 0,003. La SG des patients non greffés était de 40 % vs 84 % des patients greffés (p = 0,007). La SSR du groupe de patients non greffés et ceux greffés étaient respectivement de 40 % et 93 % (p = 0,003).
Conclusion. LAM avec caryotype normal présente un risque cytogénétique intermédiaire, et constitue un groupe hétérogène. L’indication d’un traitement intensif par AGMO a été révisée et réservée actuellement pour les patients ayant un profil mutationnel défavorable (mutation FLT 3 +). Dans notre centre et en attendant l’introduction de l’étude moléculaire, l’AGMO reste toujours proposée pour tous les patients ayant un donneur HLA compatible compte tenant des résultats satisfaisants : 1 seul cas de rechute, 1 seul cas de décès toxique avec des survies meilleures par rapport au groupe de patients n’ayant pas une AGMO.
06-47
Leucémie aiguë lymphoblastique de l’adolescent et du jeune adulte à Casablanca : profil clinicobiologique et résultats thérapeutiques
M. Dakkoune*, Q. Meryem, S. Cherkaoui, N. Khoubila, M. Lamchahab, M. Rachid, A. Madani, Q. Asmaa
Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc
Introduction. La leucémie aiguë lymphoblastique (LAL) est de très bon pronostic chez l’enfant avec un taux de guérison allant jusqu’à 90 %. Cependant chez l’adolescent et le jeune adulte (AJA) les résultats thérapeutiques sont moins bons et varient entre 80-85 %. Plusieurs facteurs ont un impact sur la réponse thérapeutique : l’hétérogénéité cytogénétique et moléculaire de la maladie (l’incidence élevée de phénotype T et de t(9;22)), les facteurs liés au patient (physiques et psychosociaux), ainsi que la durée de traitement. L’objectif de notre travail était de décrire le profil épidémiologique, clinicobiologique et évolutif de la LAL des patients âgés de 19 à 39 ans au sein de service d’hématologie-oncologie pédiatrique de Casablanca.
Patients et méthodes. Étude rétrospective descriptive étalée sur 6 ans du janvier 2012 au décembre 2017, incluant tous les patients âgés de 19 à 39 ans, suivis pour LAL dans notre service. Les données épidémiologiques, cliniques et thérapeutiques des patients ont été collectées à l’aide d’une fiche d’exploitation à partir des dossiers médicaux. L’analyse statistique a été effectuée par SPSS 18,0. Les courbes de survie ont été réalisées par la méthode non paramétrique de Kaplan Meier.
Résultats. 138 malades étaient inclus dans cette étude, l’âge médian était de 25 ans avec un sex-ratio (H/F) de 2,83, le délai médian au diagnostic était de 1 mois, le taux initial des GB était < 50 G/L chez 67 % des cas, entre 50 et 100 G/L chez 12 %, et supérieur à 100 G/L chez 21 %. Le syndrome de lyse tumorale était observé chez 11 % des malades et l’atteinte neurologique chez 10 % des malades dont 43 % étaient par une infiltration blastique du LCR. L’immunophénotypage était contributif chez 118 malades (85,5 %), 60 cas avaient un phénotype T (51 %), 52 phénotypes B (44 %) et 6 biphénotypique (4 %). Le caryotype était contributif chez 107 cas (77,5 %), il s’agissait du caryotype normal dans 32 % des cas, d’hyperdiploïde dans 14 % des cas, de la t(9;22) dans 12 % des cas, du caryotype complexe dans 6 % des cas, et d’autres anomalies dans 36 % des cas. Parmi les 138 malades, 127 étaient évaluables pour le traitement, selon le protocole thérapeutique, 118 malades (93 %) avaient reçu une chimiothérapie selon le protocole MARALL 2006 et 9 (7 %) avaient reçu le protocole GRALL 2005. Le statut postinduction était en faveur d’une RC chez 86 malades (68 %), échec chez 19 malades (15 %), 13 PDV (10 %), et 9 décès (7 %) aux cours des inductions. La rechute était survenue chez 59 malades, elle était médullaire chez 39 cas (66 %), neurologique chez 13 cas (22 %) et neurologique et médullaire chez 7 cas (12 %) avec un délai médian à la rechute de 9 mois [2-73 mois]. La SG et la SSP à 36 mois étaient respectivement de 56 % et 23 %.
Conclusion. Notre profil épidémiologique est très particulier, avec une prédominance des formes de mauvais pronostic : une incidence plus élevée de phénotype T, les formes hyperleucocytaires, les caryotypes complexes avec un taux élevé de translocation (9,22), d’où la nécessité d’une approche multidisciplinaire pour améliorer la survie et la qualité de vie de cette entité des patients.
06-48
Une nouvelle mutation germinale de PAX5 provoque une leucémie aiguë lymphoblastique B chez les adolescents et les jeunes adultes
L. Jamrog1, N. Duployez2, V. Fregona1, L. Fenwarth2, S. Lejeune3, N. Helevaut2, S. Geffroy2, A. Caillault2, A. Marceau-Renaut2, S. Poulain2, C. Roche-Lestienne4, C. Berthon5, B. Nelken6, J. Fernandes7, N. Prade8, S. Lagarde8, S. Hébrard1, C. Villenet9, M. Figeac9, B. Gerby1, E. Delabesse8, C. Preudhomme2, C. Broccardo*1
1 Équipe : altération des facteurs de transcription dans les leucémies aiguës, Centre de Recherches en Cancérologie de Toulouse - CRCT, Toulouse ; 2 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 3 Génétique clinique, CHU Lille, Lille ; 4 Institut d’hématologie, Inserm U837, CHRU Lille, Lille ; 5 Maladie du sang, CH Régional Universitaire de Lille, Lille ; 6 Hématologie pédiatrique, CH Régional Universitaire de Lille, Lille ; 7 Service d’hématologie clinique, CH De Valenciennes, Valenciennes ; 8 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 9 Plateforme de génomique fonctionnelle, IRCL, Institut pour la Recherche sur le Cancer de Lille, Lille
Introduction. PAX5 est un facteur de transcription crucial pour le maintien de l’identité des cellules lymphoïdes B. Il est la cible la plus fréquente des mutations somatiques dans les leucémies aiguës lymphoblastiques de type B (LAL-B), étant altéré dans environ 30 % des cas pédiatriques et adultes. Récemment, des mutations germinales du domaine codant l’octapeptide de PAX5 ont été décrites comme étant associées à des cas de LAL-B.
Nous détaillons ici à la fois l’identification de PAX5R38H, une mutation germinale du domaine de liaison à l’ADN de PAX5 dans une famille atteinte de LAL-B et son impact sur la différenciation B grâce un travail collaboratif entre Lille et Toulouse.
Patients et méthodes. Le Proband (II.2) est un homme de 40 ans rapidement décédé après une hémorragie intracrânienne pendant le diagnostic de LAL-B. Son passé médical a été marqué par un diagnostic antérieur de LAL-B à l’âge de 17 ans. Après une première rechute (2 ans après le diagnostic initial), il a subi un greffe de cellules souches hématopoïétiques (GCSH) allogénique d’un donneur apparenté (II.3, son frère alors âgé de 11 ans au moment de la transplantation), permettant une rémission complète (RC) pendant plus de 20 ans. Plusieurs années auparavant, sa sœur (II.1) a développé une LAL-B à l’âge de 11 ans pour laquelle elle a reçu une chimiothérapie sans GCSH. Au moment de ce résumé, elle est toujours en RC plus de 30 ans après le diagnostic initial. Enfin, le jeune frère du proband a également développé une LAL-B à l’âge de 25 ans (c.-à-d. 14 ans après avoir été donneur de GCSH du proband) et est décédé de complications infectieuses après la GCSH avec un donneur non apparenté. Les deux parents n’ont pas développé de LAL-B.
Résultats. Le séquençage de l’exome a été effectué chez le patient II.3 et a révélé une substitution germinale hétérozygote c.113G > A : p.arg38His dans PAX5. Ce variant R38H n’a pas été décrit précédemment comme un polymorphisme mais est retrouvé dans des cas rares de patients atteints de LAL-B sporadiques. PAX5R38H a été retrouvé par séquençage Sanger chez tous les sujets touchés. Cette mutation a été transmise par la mère à ses trois enfants qui ont tous développé une LAL-B à 11, 17 et 25 ans.
Afin de connaître l’impact de PAX5R38H sur la différenciation B nous l’avons étudié dans des tests de différenciation ex vivo. À l’inverse de PAX5 sauvage, PAX5R38H n’est pas capable de complémenter le défaut en Pax5 des cellules pro-B Pax5-/-. Par contre dans les cellules pro-B Pax5+/-, le mutant PAX5R38H a un effet hypomorphe restaurant de façon peu efficace la capacité de différenciation des cellules pro-B par rapport la complémentation avec PAX5 sauvage. Le transcriptome des pro-B infectées par R38H est en cours d’analyse et permettra de déterminer s’il est juste une version moins efficace de PAX5 ou s’il participe activement à la leucémogenèse en allumant un programme génique spécifique.
Conclusion. Nous décrivons la première mutation somatique du domaine de liaison à l’ADN de PAX5 chez une famille dont les trois enfants ont développé une LAL-B. Les souris Pax5+/- sont plus enclines à développer des LAL-B induites (traitement chimique, infection virale). Le mutant PAX5R38H par son activité hypomorphe semble conférer une prédisposition aux LAL-B également renforçant l’importance de PAX5 dans l’oncogenèse B étant à la fois oncogène (quand muté) ou suppresseur de tumeur. Cela montre également que le nombre de cas d’altérations germinales de PAX5 dans une famille avec LAL-B fréquentes est sous-estimé.
06-49
Une expérience monocentrique du protocole cladribine-cytarabine-filgrastim-mitoxantrone chez des patients atteints de leucémies aiguës myéloblastiques de haut risque ou en rechute/réfractaires
L. Hamri1, V. Vidal*1, A. Marceau-Renaut2, F. Sicre De Fontbrune3, V. Eclache4, N. Osman5, B. Papoular6, D. Lusina4, S. Brechignac1, R. Peffault De Latour3, C. Preudhomme2, C. Gardin1, T. Braun1
1 Service hématologie clinique, Hôpital Avicenne AP-HP, Bobigny ; 2 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 3 Service d’hématologie greffe, Hôpital Saint-Louis AP-HP, Paris ; 4 Laboratoire d’hématologie, Hôpital Avicenne, Bobigny ; 5 Unité de pharmacie clinique en oncohématologie, Hôpital Avicenne AP-HP, Bobigny ; 6 Hématologie, CHU de Nîmes (CHU), Nîmes
Introduction. Les leucémies aiguës myéloblastiques de haut risque (LAM-HR), incluant les LAM avec anomalies associées aux myélodysplasies et les LAM postchimiothérapie, sont associées à un taux de rémission complète faible et un devenir péjoratif. Il n’y a pas de standard thérapeutique concernant les LAM en rechute ou réfractaires (R/R), notamment chez les patients âgés, néanmoins conduire ces patients à l’allogreffe reste la seule stratégie curative chez les sujets éligibles. L’ajout de cladribine, un analogue des bases puriques, à la cytarabine offre une option thérapeutique. Étant donné les faibles données de la littérature, nous rapportons notre expérience chez les patients atteints de LAM-HR, en première ligne ou chez des patients R/R.
Matériels et méthodes. De janvier 2015 à juillet 2018, 20 patients consécutifs atteints de LAM-HR étaient traités dans notre centre par un cycle de CLAG-M cladribine 5 mg/m2/j (jours 2-6), cytarabine 2 g/m2/j (jours 2-6), filgrastim 300 mg/j (jours 1-6), mitoxantrone 10 mg/m2/j (jours 2-4)). Une analyse génétique avec un panel de 37 gènes a été effectuée pour 17 patients qui ont été classifiés selon Lindley et al. Blood 2015.
Résultats. 4 patients avec une LAM secondaire et 4 patients avec une LAM induite ont été traités en première ligne (5 RC, 1 décès précoce et 2 échecs), la cytogénétique ELN était adverse (n = 5), intermédiaire (n = 1) ou un échec (n = 2). Chez 12 patients antérieurement traités, la première ligne comportait une chimiothérapie intensive de type (3+7) chez 10 patients et de l’azacitidine chez 2 patients, le délai médian entre le diagnostic et le traitement par CLAG-M était de 17 (3-29) mois, la cytogénétique ELN était adverse (n = 5), intermédiaire (n = 5) ou un échec (n = 2). 7 patients ont obtenu une réponse (6 RC, 1 RCi), 3 échecs et 2 décès liés à un sepsis. 17 patients pouvaient être classés selon Lindsley et al en fonction des mutations identifiés par NGS. Les données NGS manquaient chez 3 patients présentant une cytogénétique ELN adverse. 5/7 patients présentant une LAM secondaire obtenaient une RC, 3/4 patients présentant une pan-LAM obtenaient une RC, et 2/6 patients présentant une LAM TP53+ obtenaient une RC. Au total, 12/20 patients ont obtenu une RC (incluant 1 RCi) malgré une cytogénétique adverse ou une situation de rechute. Le traitement était globalement bien toléré avec 3 décès liés à une 2 pneumonies non documentées ou 1 sepsis bactérien sans autre toxicité inhabituelle. 7 patients en RC ont reçu un deuxième CLAG avant allogreffe et 3 patients > 70 ans des traitements de consolidation moins intensifs. 15 patients étaient éligibles à une allogreffe, 8 patients obtenaient une RC et recevaient une allogreffe. Chez les allogreffés, 4 sont vivants (6 mois, 3,2 ans, 3,6 ans, 4 ans), 2 décédaient de rechute précoce et 2 décédaient de toxicité liée à la greffe.
Conclusion. L’utilisation du protocole CLAG-M, chez cette petite cohorte de patients atteints de LAM-HR, majoritairement âgés de plus de 60 ans, permet l’obtention de 60 % de RC malgré des caractéristiques hématologiques défavorables. 50 % des patients initialement éligibles à une allogreffe y ont effectivement été conduits sans toxicité inhabituelle.
06-50
Les marqueurs oncogénétiques prédictifs du devenir des patients âgés (> 60 ans) atteints de leucémie aiguë myéloïde et traités intensivement : analyse de l’essai ALFA1200
E. Fournier*1, R. Itzykson2, T. Braun3, C. Berthon4, A. Marceau-Renaut5, C. Pautas6, E. Lemasle7, P. Turlure8, N. Duployez9, JB. Micol10, L. Ades11, JP. Marolleau12, L. Gastaud13, P. Rousselot14, X. Thomas15, S. Chantepie16, C. Terré17, H. Dombret18, C. Gardin19, C. Preudhomme9
1 Laboratoire d’hématologie, CH Régional Universitaire de Lille, Lille ; 2 Hématologie, Hôpital Saint-Louis, Paris ; 3 Hématologie, Hôpital Avicenne, Bobigny ; 4 Maladie du sang, CH Régional Universitaire de Lille, Lille ; 5 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 6 Hématologie clinique et thérapie cellulaire, Hôpital Henri Mondor, Créteil ; 7 Département d’hématologie, Hôpital Henri Becquerel, Rouen ; 8 Hématologie, CHU Limoges, Limoges ; 9 Laboratoire d’hématologie, CHU Lille, Lille ; 10 Service d’hématologie clinique, Gustave Roussy, Villejuif ; 11 Hématologie seniors, Hôpital Saint-Louis, Paris ; 12 Maladies du sang, Chu Amiens, Amiens ; 13 Oncologie, Centre Antoine Lacassagne, Nice ; 14 Hématologie et Oncologie, CH de Versailles, Versailles ; 15 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 16 Service d’hématologie clinique, CHU Caen, Caen ; 17 Laboratoire de cytogénétique, Centre Hospitalier de Versailles, Le Chesnay ; 18 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 19 Service d’hématologie clinique, Hôpital Avicenne, Bobigny
Introduction. Les LAM sont un groupe d’hémopathies hétérogènes. Les anomalies récurrentes sont essentielles à la stratification pronostique. Les classifications utilisées ont été établies à l’aide de cohorte de patients jeunes. Cependant, des interactions majeures peuvent survenir avec l’âge conduisant à une valeur pronostique incertaine de ces mutations récurrentes.
Patients et méthodes. Les échantillons au diagnostic de LAM de patients participant à l’étude ALFA1200 ont été séquencés PAR NGS à l’aide d’un panel de 37 gènes selon la technique Proton ThermoFisher.
Résultats. Le séquençage a été réalisé pour 471 patients inclus dans l’étude. L’âge médian est de 68 ans. Le risque selon l’ELN 2017 est favorable, intermédiaire, et défavorable dans 3 %, 78 % et 19 % respectivement. La RC a été atteinte chez 72 % patients. Avec un suivi médian de 25,4 mois, la valeur médiane de l’OS est de 20,7 mois. Le taux de RC des patients présentant une LAM à risque génétique fav, int et def est de 85 %, 76 % et 57 % respectivement. Les patients présentent une médiane de 3 mutations. Les anomalies les plus fréquemment rencontrées sont DNMT3A (29 %), NPM1 (27 %), TET2 (21 %), ASXL1 (19 %), FLT3-ITD (19 %), SRSF2 (18 %), IDH2 (18 %), RUNX1 (17 %), NRAS (13 %) et IDH1 (10 %). En analyse univariée, dans le groupe cytogénétique int et fav (n = 355), les mutations d’IDH2R140, NPM1 et DNMT3A prédisent un taux de RC supérieur (p < 0,05). Inversement, les mutations de RUNX1, ASXL1, TET2 et NRAS (p < 0,01) étaient associées à un taux inférieur de mise en RC. En analyse multivariée, seule la mutation de NPM1 améliore le taux de RC, tandis que les mutations d’ASXL1, RUNX1 et NRAS conservent leur valeur prédictive négative. En analyse multivariée sur l’OS, les mutations de NPM1 (p < 0,001) et SRSF2 (p < 0,05) sont de meilleur pronostic au contraire des mutations DNMT3A (p < 0,001), ASXL1, SRSF2 (p < 0,01) et FLT3-ITD (allèle ratio ≥ 0,5) (p < 0,05). Parmi elles, des interactions significatives ont été trouvées entre NPM1, SRSF2 et DNMT3A. Dans le groupe cytogénétique def, seule la mutation de TP53 conduit à une diminution significative de l’OS. Ceci a conduit à redéfinir une hiérarchie pronostique pour les patients âgés en fonction de leurs anomalies moléculaires.
Conclusion. Dans les LAM du sujet âgé, la recherche de mutations dans 7 gènes (NPM1, SRSF2, FLT3, DNMT3A, ASLX1, NRAS et TP53) peuvent améliorer la stratification pronostique.

06-51
L’absence de blastes chez les corticosensibles est-elle prédictive de meilleures survies au cours des leucémies aiguës lymphoblastiques de l’enfant ?
I. Frikha*, I. Turki, M. Medhaffer, S. Hadijji, H. Bellaaj, O. Kassar, M. Ghorbel, I. Ben Amor, F. Kallel, M. Charfi, M. Elloumi
Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie
Introduction. De nombreux facteurs pronostiques ont été identifiés au cours de ces dernières décennies dans la leucémie aiguë lymphoblastique (LAL) de l’enfant. La résistance initiale au traitement, observée au cours de l’induction (corticorésistance et/ou chimiorésistance), est un facteur pronostique péjoratif. Dans cette étude, nous avons analysé l’impact pronostique de la persistance des blastes au 8e jour de préphase chez les patients corticosensibles.
Patients et méthodes. Nous avons analysé rétrospectivement les données de 264 enfants et adolescents traités selon le protocole EORTC 58 951 durant la période de janvier 2000 à décembre 2015. Nous avons particulièrement analysé l’impact pronostique de la corticosensibilité (Cs), définie par un taux de blastes sanguins < 1 000 blastes/mm3 après 7 jours de dexaméthasone à la dose de 3 mg/m2/12 h et une injection intrathécale de méthotrexate seul (préphase). Nous avons analysé, dans le groupe des Cs, les résultats de 2 sous-groupes individualisés : le premier avec 0 blaste à J8 de la préphase (Cs = 0) et le deuxième avec présence de blastes (Cs ǂ 0).
Résultats. Nous avons colligé 264 cas, 192 enfants et 72 Adolescents. La médiane d’âge était de 10 ans. Le sex-ratio était de 1,5. Il s’agit d’une LAL de phénotype B dans 64 % des cas et de phénotype T dans 35 % des cas. Les patients ont été stratifiés en groupes de risque faible dans 11 cas (4 %), risque moyen dans 182 cas (68 %) et risque élevé dans 71 cas (27 %). Deux cent neuf patients (79 %) étaient corticosensibles dont 150 patients (28 %) n’avaient pas de blastes à J8 de la préphase. Les résultats thérapeutiques de notre série dans les 2 sous groupes Cs = 0 et Cs ǂ 0 sont représentés dans le tableau suivant :Dans toute la série Pour les corticosensibles Corticosensible Corticorésistant p Cs = 0 Cs ǂ 0 p RC 93 % 82 % 0,6 95 % 89 % 0,31 Rechutes 27 % 38 % 0,12 18 % 51 % < 10-5 SG à 5 ans 68 % 35 % < 10-5 73 % 55 % 0,061 SSE à 5 ans 63 % 35 % < 10-4 69 % 47 % 0,005 SSM à 5 ans 72 % 52 % 0,004 82 % 50 % < 10-5
Conclusion. Le rôle pronostique de la corticosensibilité a été validé par plusieurs essais successifs et constitue le premier élément d’évaluation précoce de la réponse au traitement. Encore plus, nous avons démontré dans notre étude que l’absence vs la présence de blastes an frottis sanguin de J8 de préphase chez les patients corticosensibles constitue un paramètre discriminant aussi bien pour la survenue de rechute que pour la survie. Ce paramètre doit être validé à grande échelle pour pouvoir intensifier rapidement chez les mauvais répondeurs à la préphase.
06-52
Leucémie aiguë myéloïde postchimiothérapie avec t(8;16) : un nouveau cas atypique et revue de la littérature
M. Haas*1, K. Mheidly2, S. Toujani3, C. Pastoret1, J. Goustille1, C. Henry3, M. Bernard2, B. Ly Sunnaram1, T. Fest1
1 Laboratoire d’hématologie, CHU Rennes - Hôpital Pontchaillou, Rennes ; 2 Hématologie clinique, CHU Rennes - Hôpital Pontchaillou, Rennes ; 3 Laboratoire de cytogénétique et biologie cellulaire, CHU Rennes - Hôpital Pontchaillou, Rennes
Introduction. Les LAM post-chimiothérapie (t-AML) représentent environ 10 % des cas de LAM. La translocation t(8;16) est une anomalie rare, retrouvée dans environ 0,5 % des LAM qui sont soit de novo ou de type t-AML. Ces leucémies sont associées à un profil clinique, morphologique et génétique spécifique, ainsi qu’à un mauvais pronostic. Cette translocation chromosomique conduit à la fusion du gène KAT6A (8p11) avec le gène CREBBP (16p13), tous deux codant pour des histones acétyltransférases.
Patients et méthodes. Nous présentons le cas d’une femme de 74 ans développant une LAM après 17 mois de rémission complète pour un lymphome centroblastique B traité par R-CHOP. Curieusement, les blastes leucémiques présentaient des caractéristiques mimant une leucémie aiguë promyélocytaire (LAP). Les analyses génétiques et moléculaires de la LAM notaient l’absence de transcrits PML-RARA, la présence d’une translocation t(8;16)(p11;p13), et la mise en évidence d’un remaniement impliquant le gène KATA6. Une analyse NGS a permis de détecter une mutation de TET2 (VAF = 27 %). Cette observation nous a poussés à étudier les autres cas de t-AML avec t(8;16) rapportés dans la littérature et d’envisager à la lumière des connaissances actuelles les hypothèses physiopathologiques qui pourraient sous-tendre une telle entité.
Résultats. Selon la classification FAB, ces leucémies sont classées en LAM-M4, M5a et M5b. Cependant, un pléomorphisme cytologique pouvant évoquer un diagnostic de LAP est fréquemment rapporté, suggérant que la LAM avec t(8;16) provient d’une cellule souche très précoce avec un potentiel de différenciation myéloïde et monoblastique. À ce jour, 49 cas de t-LAM avec t(8;16) ont été rapportés dans la littérature, et particulièrement suite à des chimiothérapies incluant les anthracyclines. Les gènes KAT6A et CREBBP pourraient alors être une cible privilégiée des anthracyclines. Chez notre patiente nous avons identifié une mutation TET2 ce qui pose la question de son occurrence par rapport à la t(8;16) sachant que la mutation de ce gène est souvent liée à une hématopoïèse clonale qui pourrait être éventuellement associée avec le développement du lymphome initial. Afin d’avancer dans ces hypothèses nous envisageons d’effectuer une analyse NGS plus étendue des tumeurs en comparant leurs profils à la recherche d’atteintes somatiques communes dont TET2. La translocation t(8;16) génère un gène de fusion KAT6A-CREBBP qui conserve une activité histone acétyltransférase. Ce point pourrait expliquer la spécificité du profil d’expression génique décrite pour cette entité comparée aux autres LAM, ce profil ayant de grandes similitudes avec celui des LAM avec réarrangement MLL, et inversement, est bien différent de celui des LAM CBF et des LAP. Cela suggère donc une voie commune de leucémogenèse.
Conclusion. La t-AML avec t(8;16) est une entité rare et distincte des autres LAM. L’implication de gènes particuliers de l’épigénome dans cette LAM ouvre la possibilité d’envisager des stratégies thérapeutiques nouvelles avec des médicaments agissant sur l’épigénome et aussi probablement vers des molécules qui favoriseraient la mort cellulaire par apoptose en bloquant BCL2 (Vénétoclax).
Dans notre présentation, nous discuterons les différents versants du diagnostic biologique ainsi que les hypothèses physiopathologiques émises, et aussi, nous ferons part de nos résultats biologiques actuellement en attente.
06-53
Apports respectifs du caryotype et de l’hybridation in situ fluorescente dans les leucémies aiguës myéloblastiques de novo de l’adulte
S. Taoussi*, F. Lamraoui, S. Oukid, N. Rekab, KM. Benlabiod, Y. Bouchakor Moussa, MT. Abad, M. Bradai
Hématologie, CAC, Laboratoire Hémopathies malignes et hémoglobinopathies, Université Blida 1, Blida, Algérie
Introduction. Les anomalies cytogénétiques récurrentes et non aléatoires sont présentes dans 50-60 % des leucémies aiguës myéloblastiques. La classification cytogénétique des LAM (OMS 2016) stratifie ces entités en 3 groupes pronostiques, le groupe favorable [t (15 ; 17) (q24 ; q21), t(8;21)(q22;q22), inv(16)(p13,1q22) ou t(16;16)(p13,1;q22)], le groupe intermédiaire [caryotype normal, t(9;11)(p21;q23), autres anomalies non classées comme favorables ou défavorables] et le groupe défavorable [caryotype complexe (≥ 3 anomalies), monosomie 5/del(5q), monosomie 7/del(7q), anomalies 17p, t(v;11)(v;q23) avec réarrangement KMT2A (excepté la t(9;11)(p21; q23)), t(6;9)(p23;q34) (gène de fusion DEK-NUP214), inv(3)(q21;q26,2) ou t(3;3)(q21;q26,2)] Ces anomalies sont traditionnellement recherchées par l’étude du caryotype seul. Quelle est la place de la cytogénétique moléculaire (FISH) dans l’étude des LAM de novo ?
Patients et méthodes. Objectif : Préciser l’apport respectif du caryotype et de la FISH pour la révélation des anomalies chromosomiques dans les leucémies aiguës myéloblastiques de novo.
Dans 108 cas de LAM de novo, diagnostiquées par cytologie sanguine et médullaire et dans certains cas par cytométrie en flux, nous avons pratiqué systématiquement un caryotype en bandes R et une FISH en utilisant les sondes suivantes : t(8;21) (RUNX1-RUNX1T1), KMT2A (11q23), inv(16)/t(16;16) (CBFB/MYH11), PML/RARA ; RAR break pour les LAP, la recherche du bcr-abl et de la del(P53). Pour les types M0 et M indifférenciées, les sondes 5q31,1, 7q22-q31 et 20q- ont été appliquées.
Résultats. Un échec du caryotype a été noté dans 11 cas (10 %). Le caryotype interprétable (n = 97) était normal dans 35 cas (36 %). Dans 62 cas (64 %) il a retrouvé 8 inv(16), 8 t(8;21), 8 t(15;17), 9 hyperdiploïdies, 10 cas de trisomies, 5 monosomies, 3 caryotypes complexes, 4 caryotypes monosomal, 5 cas de translocations rares [t(1;14)(p34;q32), t(4;12)(q12;p13), t(16;21)(p11;q22), t(9;22)(q34;q11/slx2, t(6;11)(q27;q23)], 1 cas avec perte de l’Y et un cas avec iso21q.
Dans 35 cas où le caryotype était normal, 2 inv(16), 4 cas de réarrangement KMT2A ont été retrouvés par la FISH ; dans 11 cas d’échec du caryotype, la FISH a retrouvé : 4 cas avec t(8;21), un cas avec inv(16), une del(20q) et une del(21q).
Dans 7 cas où la FISH n’a pas montré d’anomalies avec les sondes utilisées, le caryotype a objectivé : -Y : 1 cas, -19 : 1 cas, t(16;21)(p11;q22) : 1 cas, t(1;14)(p34;q32) : 1 cas, t(4;12)(q12;p13) : 1 cas.
La stratification en groupes pronostiques a donné : groupe favorable (n = 31) (29 %), groupe intermédiaire (n = 56) (51,5 %), groupe défavorable (n = 21) (19,5 %).
Conclusion. Le caryotype est essentiel à l’évaluation pronostique des LAM. La FISH, complète utilement le caryotype lorsqu’existent des anomalies cryptiques ou en cas d’échec de ce dernier, comme cela a été montré dans notre approche. La répartition selon les groupes pronostiques montre dans notre travail une proportion de cas rejoignant les données des séries du CALGB (2002) et marocaine (2009). Nos données doivent être évaluées sur une cohorte plus importante de patients. Ainsi, l’utilisation concomitante du caryotype et de la FISH permet d’optimiser la détection des anomalies génomiques dans les LAM de novo à des fins diagnostiques, thérapeutiques et pronostiques.
06-54
Résultats chez des patients âgés présentant une LAM de mauvais pronostic/secondaire récemment diagnostiquée qui atteignent une rémission après induction par CPX-351 ou par le schéma 7+3
S. Faderl1, GJ. Schiller2, JE. Cortes3, G. Guindeuil*4, RJ. Ryan5, M. Chiarella1
1 Department of clinical development, Jazz Pharmaceuticals, Palo Alto, CA, États-Unis ; 2 David geffen school of medicine, University of California – Los Angeles, Los Angeles, CA, États-Unis ; 3 Department of leukemia, University of Texas MD Anderson Cancer Center, Houston, TX, États-Unis ; 4 French medical officer, Jazz Pharmaceuticals, Lyon ; 5 Department of biostatistics, Jazz Pharmaceuticals, Palo Alto, CA, États-Unis
Introduction. CPX-351 (Vyxeos®) est une formulation liposomale de deux médicaments co-encapsulés, la cytarabine et la daunorubicine, selon un ratio synergique, et est approuvé par l’EMA et la FDA pour les adultes présentant une LAM secondaire à un traitement ou une LAM avec anomalies associées aux myélodysplasies. Cette analyse post hoc des données d’une étude de phase III a évalué les résultats chez des patients âgés de 60 à 75 ans présentant une LAM de mauvais pronostic/ LAMs récemment diagnostiquée et obtenant une rémission après induction par CPX-351 ou par schéma 7+3 (cytarabine + daunorubicine).
Patients et méthodes. Patients randomisés selon un rapport 1:1 pour recevoir jusqu’à 2 inductions de CPX-351 (100 unités/m2 [100 mg/m2 de cytarabine + 44 mg/m2 de daunorubicine] les jours 1, 3 et 5 [deuxième induction : jour 1 et 3]) ou du schéma 7+3 (100 mg/m2/jour de cytarabine sur 7 jours [deuxième induction : 5 jours] + 60 mg/m2 de daunorubicine les jours 1 à 3 [deuxième induction : jour 1 et 2]). Les patients obtenant une réponse complète (RC) ou une RC avec récupération incomplète de la numération plaquettaire ou des neutrophiles (RCi) pouvaient recevoir jusqu’à 2 cycles de consolidation.
Résultats. CPX-351 était associé à des taux significativement plus élevés que le schéma 7+3 de RC + RCi (73/153 [47,7 %] contre 52/156 [33,3 %] ; P bilatéral = 0,016) et RC (57/153 [37,3 %] contre 40/156 [25,6 %] ; p bilatéral = 0,040). Survie globale (SG) médiane plus longue avec CPX-351 qu’avec le schéma 7+3 chez les patients ayant obtenu une RC + RCi (25,43 contre 10,41 mois ; RR = 0,49 [IC à 95 % : 0,31-0,77]) ou une RC (25,43 contre 10,97 mois ; RR = 0,49 [IC à 95 % : 0,29-0,83]). Parmi les patients présentant une RC + RCi, 40/73 (55 %) dans le bras CPX-351 et 24/52 (46 %) dans le bras 7+3 ont reçu une greffe ; SG médiane calculée à partir de la date de la greffe était non atteinte contre 11,65 mois, respectivement (RR = 0,42 [IC à 95 % : 0,20-0,86]). Les EI graves chez ≥ 5 % des patients présentant une RC + RCi étaient neutropénie fébrile (CPX-351 : 15 % ; 7+3 : 12 %), insuffisance respiratoire aiguë (7 % ; 2 %), diminution de la fraction d’éjection (5 % ; 4 %), septicémie (5 % ; 4 %), pneumonie (3 % ; 6 %) et œdème pulmonaire (1 % ; 6 %). Décès de quatre patients présentant une RC + RCi pendant la phase de traitement (septicémie [CPX-351] ; fibrillation atriale [7+3] ; progression de la maladie [7+3] ; hémorragie intracrânienne [7+3]) ; aucun patient des deux bras n’a présenté de mortalité précoce à 60 jours. Chez les patients présentant une RC + RCi, le délai médian de récupération de la numération des neutrophiles ≥ 1,000/μL et de la numération plaquettaire ≥ 100 000/μL était plus long avec CPX-351 (37 et 42 jours) qu’avec le schéma 7+3 (29 et 32 jours).
Conclusion. CPX-351 était associé à une SG plus longue chez les patients ayant obtenu une RC + RCi, suggérant une réponse potentiellement plus profonde et le profil de sécurité était conforme à celui du schéma 7+3.
06-55
Résultats chez des patients âgés présentant une leucémie aiguë myéloïde nouvellement diagnostiquée de mauvais pronostic/secondaire qui ont reçu une consolidation dans une étude de phase III évaluant le CPX-351 par rapport au schéma conventionnel 7+3/5+2
JE. Kolitz1, JE. Cortes2, D. Hogge3, G. Guindeuil*4, RJ. Ryan5, M. Chiarella6
1 -, Monter Cancer Center, Northwell Health System, Lake Success, NY, États-Unis ; 2 Department of leukemia, University of Texas MD Anderson Cancer Center, Houston, TX, États-Unis ; 3 -, Leukemia/BMT Program of British Columbia, Vancouver, BC, Canada ; 4 French medical officer, Jazz Pharmaceuticals, Lyon ; 5 Department of biostatistics, Jazz Pharmaceuticals, Palo Alto, CA, États-Unis ; 6 Department of clinical development, Jazz Pharmaceuticals, Palo Alto, CA, États-Unis
Introduction. CPX-351 (Vyxeos®) est une formulation liposomale de deux médicaments co-encapsulés, la cytarabine et la daunorubicine, selon un ratio synergique, et est approuvé par l’EMA et la FDA pour les adultes présentant une LAM nouvellement diagnostiquée secondaire à un traitement ou une LAM avec anomalies associées aux myélodysplasies. Cette analyse post hoc des données d’une étude de phase III chez des adultes âgés de 60 à 75 ans présentant une LAM nouvellement diagnostiquée de mauvais pronostic/ LAMs a évalué l’impact sur les résultats d’une consolidation par CPX-351 ou par le schéma 5+2 associant la cytarabine et la daunorubicine.
Patients et méthodes. Patients randomisés selon un rapport 1:1 pour recevoir jusqu’à 2 inductions de CPX-351 (100 unités/m2 [100 mg/m2 de cytarabine + 44 mg/m2 de daunorubicine] les jours 1, 3 et 5 [deuxième induction : les jours 1 et 3]) ou du schéma 7+3 / 5+2 (100 mg/m2/jour de cytarabine pendant 7 jours [deuxième induction : 5 jours] + 60 mg/m2 de daunorubicine les jours 1 à 3 [deuxième induction : les jours 1 et 2]). Les patients obtenant une réponse complète (RC) ou une RC avec récupération incomplète de la numération plaquettaire ou des neutrophiles (RCi) pouvaient recevoir jusqu’à 2 consolidations de CPX-351 (65 unités/m2 [65 mg/m2 de cytarabine + 29 mg/m2 de daunorubicine]) les jours 1 et 3 ou 5+2 (comme en deuxième induction).
Résultats. L’étude a inclus 309 patients (CPX-351 : n = 153 ; 7+3 : n = 156). La SG médiane était de 9,56 mois avec le CPX-351 et de 5,95 mois avec le schéma 7+3 (RR = 0,69 [IC à 95 % : 0,52-0,90] ; P unilatéral = 0,003) ; une RC + RCi a été obtenue par 73 (47,7 %) contre 52 (33,3 %) patients, respectivement. La survie médiane pour les patients ayant par la suite reçu une consolidation (CPX-351 : n = 49 ; 7+3 : n = 32) était plus longue avec le CPX-351 que le schéma 7+3 / 5+2 (25,43 contre 8,53 mois ; RR = 0,44 [IC à 95 % : 0,25-0,77]), que les patients aient reçu une consolidation suivie d’une greffe (non atteinte [n = 24] contre 9,82 mois [n = 12] ; RR = 0,28 [0,11-0,72]) ou non suivie d’une greffe (13,67 [n = 24] contre 8,44 mois [n = 20] ; RR = 0,62 [0,31-1,26]). Le pourcentage de patients présentant des EI de grade 3 à 5 pendant la consolidation était de 53,1 % avec le CPX-351 et de 46,9 % avec 7+3/ 5+2 ; les EI de grade 3 à 5 les plus fréquents étaient la neutropénie fébrile (28,6 % contre 25,0 %), la pneumonie (8,2 % contre 6,3 %) et la cellulite (8,2 % contre 3,1 %). Le délai médian de récupération de ≥ 500 neutrophiles/μL était de 35,5 jours avec CPX-351 et de 33,5 jours avec le schéma 7+3 / 5+2 et le délai médian de récupération de ≥ 50 000 plaquettes/μL était de 37 jours avec CPX-351 et de 30 jours avec le schéma 7+3 / 5+2.
Conclusion. Un traitement continu par CPX-351 pendant l’induction et la consolidation a amélioré la SG par rapport au schéma 7+3 / 5+2 avec un profil de sécurité comparable chez les patients âgés présentant une LAM de mauvais pronostic / LAMs.
06-56
Résultats thérapeutiques du protocole EORTC 58 951 dans les leucémies aiguës lymphoblastiques de l’enfant de haut risque
I. Frikha*1, I. Turki1, M. Medhaffer1, N. Benabdeljalil2, S. Hadijji1, I. Ben Amor1, O. Kassar1, M. Ghorbel1, F. Kallel1, M. Charfi1, T. Benothmen2, M. Elloumi1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Hématologie clinique, Centre National de Greffe de la Moelle Osseuse, Tunis, Tunisie
Introduction. Plus de 80 % des leucémies aiguës lymphoblastiques de l’enfant peuvent être actuellement guéries grâce à une meilleure stratification en groupe de risque. Cependant, les résultats de certains groupes thérapeutiques dits de haut risque ou risque élevé (RE) sont non satisfaisants. L’objectif de ce travail est d’étudier les caractéristiques diagnostiques et les résultats thérapeutiques des LAL de l’enfant traités selon le bras RE du protocole EORTC 58 951.
Patients et méthodes. Entre janvier 2000 et décembre 2015, 192 cas de LAL de l’enfant âgés de moins de 16 ans ont été suivis au service d’hématologie de Sfax et traités selon le protocole EORTC58951. Quarante-sept patients ont été traités selon le groupe RE. Ce groupe RE comporte les LAL avec mauvaise réponse à la préphase (blastes > 1 000/mm3 au frottis sanguin de J8), ceux avec des anomalies cytogénétiques de mauvais pronostic (t (9,22), t(4;11), anomalies du 11q23.) ou en cas d’absence de rémission cytologique et/ou cytogénétique après 1 cure d’induction. Nous avons analysé leurs caractéristiques et les résultats thérapeutiques obtenus par ce protocole.
Résultats. Nous avons colligé 47 cas de LAL RE soit 24 % de toutes les LAL diagnostiquées durant la période d’étude. L’âge médian était de 9 ans. Le taux médian de leucocytose était de 47 G/L. Il s’agit de LAL de phénotype B dans 51 % des cas et de phénotype T dans 49 %. Au terme de la préphase, 70 % des cas étaient corticorésistants. Les critères de classification en RE étaient la présence d’anomalie cytogénétique défavorable pour 7 patients (15 %), la corticorésistance pour 30 patients (64 %) et l’échec de rémission complète (RC) à 1 cure pour 10 cas (21 %). La RC était obtenue chez 41 patients, soit un taux de RC 87 % (68 % après 1 cure). Nous n’avons noté aucun décès durant l’induction et 3 patients sont décédés au cours du traitement de consolidation. Le typage HLA a été réalisé pour 36 patients parmi les 39 ayant l’indication d’allogreffe de moelle en RC1 (typage non fait pour décès précoce = 1 patient et mauvaises conditions socio-économiques = 2 cas). Parmi ces 36 patients, 19 disposaient d’un donneur HLA compatible. Seulement 8 patients ont été allogreffés soit 20 % des patients en RC ; 7 patients d’entre eux sont vivants en RC et 1 patient est décédé par réactivation CMV postgreffe. Parmi les 33 patients en RC et non allogreffés, 16 sont vivants en RC (48 %), 3 sont décédés en cours de chimiothérapie de consolidation et 14 ont rechuté (42 %). La SSE à 5 ans de nos patients traités selon le bras RE était 50 %, elle était meilleure pour les patients allogreffés avec 87 % vs 41 % (p = 0,037).
Conclusion. Le pourcentage des LAL classées RE de 24 % dans notre population est nettement supérieur à ceux rapportés par la littérature (< 15 %). Ceci est du, entre autres, à une fréquence élevée de corticorésistance (70 % vs 10 % dans la littérature). Malgré un taux de RC de 87 %, comparable à ceux de la littérature (90 %), nous avons noté un taux de rechute de 42 % plus élevé que la littérature (20 %) et une SSE à 5 ans de 50 % inférieure à la littérature (60 à 80 %). La SSE est meilleure en cas de réalisation de l’allogreffe de moelle aussi bien dans notre série que les séries de la littérature. Nos résultats pourraient être améliorés par l’élargissement des possibilités d’allogreffe de moelle osseuse en RC1 (phéno-identique et sang de cordon).
06-57
Caractéristiques épidémiologique, clinique, biologique, thérapeutique et de survie des leucémies aiguës à promyélocytes : étude unicentrique d’une série de 43 patients
F. Soltani*, S. Refis, M. Aiche, R. Nacib, M. Merrouche, B. Garah, A. Bekache, M. Saidi
Hématologie, et Thérapie Cellulaire, CLCC Batna. Université Batna 2, Batna, Algérie
Introduction. La leucémie aiguë promyélocytaire (LAP) est un sous-type de leucémie aiguë myéloïde (LAM), représentant 5 à 15 % d’entre elles. Elle revêt de nombreuses singularités qu’il convient de souligner :
– elle est due à une anomalie cytogénétique t (15 ; 17) retrouvée dans 95 % des cas.
– elle constitue une urgence de prise en charge en raison de la coagulopathie quasi constante au diagnostic à l’origine d’une mortalité précoce initiale.
Mais une fois l’obstacle de la CIVD surmonté, le pronostic est transformé grâce à l’utilisation d’agents différenciateurs : le premier est un dérivé rétinoïque ATRA et le dernier un dérivé de l’arsenic l’ATO.
Patients et méthodes. Il s’agit d’une étude rétrospective sur une période de 10 ans (janvier 2009-décembre 2018), durant laquelle tous les cas de LAP sont répertoriés, cette étude porte sur les aspects épidémiologiques, cliniques, biologiques, thérapeutiques et évolutifs.
Résultats. Durant cette décennie, 240 LAM sont diagnostiquées dont 43 LAP (17,9 %) : 5 enfants (8-18 ans) et 38 adultes. L’âge médian des 38 adultes est de 35 ans (18-81 ans) et le sex-ratio de 1,23 (21 H/17 F). On retrouve chez 8 pts la notion de cancer familial dont 4 hémopathies malignes.
Le mode de début est représenté par un syndrome hémorragique chez 15 pts (34,88 %), une anémie chez 23 pts (53,4 %) et infection pour 5 pts.
L’examen clinique initial retrouve un syndrome hémorragique dans 32 cas (74,41 %), infections pour 16 pts (37,2 %), le syndrome tumoral est retrouvé chez 8 pts.
Sur le plan biologique, le taux de GB médian est 3 700/mm3 (400-165 000), la forme leucopénique (GB < 4 000/mm3) est rencontrée chez 22 pts (51,16 %), l’hyperleucocytose > 10 000/mm3 chez 15 pts (34,88 %) et > 50 000/mm3 chez 7 pts. Une thrombopénie < 40 000/mm3 pour 32 pts (65,5 %) et le taux d’Hb moyen est de 8,16 g/dL (4,1-12 g) dont 7 ont une anémie profonde Hb < 7 g/dL.
L’étude immunophénotypique a été réalisée uniquement pour 11 pts, aucune analyse moléculaire ou cytogénétique.
Selon le score pronostique de Sanz : 5 pts (11,62 %) sont de bas risque, 23 (53,48 %) de risque intermédiaire et 15 pts de haut risque (34,8 %).
Tous les patients ont reçu l’ATRA plus ou moins une chimiothérapie, dont 27 sont encore vivants (62,79 %) et 16 patients (37,2 %) décédés, dont 10 avant ou durant l’induction soit une mortalité précoce de 27,7 %, souvent suite à un syndrome hémorragique (7 étaient hyperleucocytaires et 5 avaient plus de 70 ans), et 6 décès tardifs : 3 après la deuxième consolidation d’infections. Seuls 2 pts ont rechuté et un décédé.
Conclusion. Les LAP représentent 15 % des LAM en Algérie ; dans cette série 43 pts, majoritairement adulte, l’âge médian est de 35 ans, la présentation initiale est le plus souvent un syndrome hémorragique. Sur le plan biologique, la forme leucopénique est de 51,16 % et la forme hyperleucocytaire de 34,88 %. Sur le plan pronostique, les haut risque représentent 3 4,8 % des cas. La mortalité précoce est de 27,7 % due le plus souvent à la CIVD qui gagne à être réduite vu les résultats obtenus après traitement.
06-58
Recherche de la maladie résiduelle par cytométrie en flux dans le sang périphérique chez les patients atteints de leucémie aiguë myéloblastique
C. Guénot1, F. Lacombe2, K. Allou2, F. Dumezy3, J. Feuillard4, F. Genevieve5, E. Guérin4, J. Guy6, M. Maynadié6, O. Wagner-Ballon7, C. Preudhomme3, A. Baruchel8, H. Dombret9, N. Ifrah10, MC. Béné*11, GEIL
1 Hématologie biologie, CH - Le Mans, Le Mans ; 2 Hématologie biologique, CHU Haut-Lévêque, Bordeaux ; 3 Laboratoire d’hématologie, CH Régional Universitaire de Lille, Lille ; 4 Hématologie Biologique, CHU Limoges, Limoges ; 5 Hématologie biologique, Centre hospitalier universitaire d’Angers, Angers ; 6 Hématologie, CHU Dijon, Dijon ; 7 Département Hématologie et Immunologie Biologiques, Hôpital Henri Mondor, Créteil ; 8 Hématologie et immunologie pédiatrique, hôpital Robert Debré, AP-HP, Paris ; 9 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 10 Service des maladies du sang, CHU - CHU Angers, Angers ; 11 Service d’hématologie biologique, Hôtel-Dieu, Nantes
Introduction. Parmi les questions en suspens concernant les stratégies de recherche de la maladie résiduelle (MRD) dans le suivi des patients atteints de leucémie aiguë myéloblastique (LAM), une des questions récurrentes concerne l’intérêt de sa recherche dans le sang périphérique des patients, en particulier par cytométrie en flux (CMF). Très peu d’études dans la littérature se sont intéressées à cette problématique, avec des résultats intéressants mais concernant peu de patients.
Patients et méthodes. Pour 96 patients suivis dans huit centres français, 217 échantillons de sang périphérique ont été analysés avec un panel restreint explorant essentiellement l’expression des marqueurs CD34, CD117, CD33 et CD13 avec un back-gating sur l’histogramme CD45/SSC et l’apport additionnel éventuel de l’expression de CD15 et CD7. Tous les listmodes de CMF aux points de suivis ont été comparés à celui du diagnostic du même patient, selon une stratégie publiée. Les acquisitions en CMF ont concerné des échantillons traités avec un protocole de « lyse sans lavage » permettant de considérer l’ensemble des leucocytes. Les recommandations étaient d’acquérir au moins 250 000 événements afin d’atteindre une sensibilité de 5×10-5. Les patients étaient traités de façon conventionnelle, au sein de protocoles de chimiothérapie conventionnels ou selon ces schémas. Les résultats ont été analysés en fonction des données de survie sans progression et de survie globale.
Résultats. Les 217 échantillons se répartissaient en 66 points post-induction, 55 points avant consolidation, 33 en fin de traitement et 61 au-delà. L’analyse a pris en considération d’une part les patients pour lesquelles une MRD n’a jamais été détectée et d’autre part les patients présentant au moins un point de MRD détectable. Ceci a permis de confirmer l’avantage en survie des patients « toujours négatifs » avec une médiane de survie non atteinte vs. 10 mois pour les patients avec MRD détectable (95 % CI 5-17, p < 0,0001). Le même résultat a été observé en ne prenant en compte que les 66 points de fin d’induction (MRD1p < 0,0001).
Conclusion. Cette étude multicentrique confirme l’intérêt d’un suivi de la MRD dans le sang périphérique en CMF chez les patients atteints de LAM. Au-delà des points très précoces perinduction, déjà rapportés dans la littérature, cette étude indique qu’une MRD positive dans le sang périphérique devrait faire reconsidérer un renforcement thérapeutique, alors que seule une MRD périphérique négative nécessiterait un contrôle médullaire.
06-59
Sarcome mégacaryoblastique avec translocation t (1;22;5) RBM15-MKL1
C. Mayeur Rousse*1, SL. Mahi2, A. Spiegel2, N. Weingertner3, MP. Chenard3, F. Gabor4, C. Gervais1, A. Eischen1, N. Guenova2, L. Mauvieux1, C. Paillard2
1 Laboratoire d’hématologie, CHRU Hôpitaux Universitaires Strasbourg, Strasbourg ; 2 Hématopédiatrie, Hôpital de Hautepierre, Strasbourg ; 3 Service de pathologie-histologie, CHRU Hôpitaux Universitaires Strasbourg, Strasbourg ; 4 Radiologie, Hôpital de Hautepierre - Hôpitaux Universitaires de Strasbourg, Strasbourg
Introduction. Les leucémies aiguës mégacaryoblastiques (LAM) avec translocation t(1;22)(p13,3;q13,1) sont des LAM pédiatriques rares, représentant moins de 1 % des cas. Nous rapportons le cas d’un enfant de 18 mois, de présentation uniquement sarcomateuse, ganglionnaire et osseuse.
Résultats. Le petit G., 18 mois, consulte une première fois aux urgences pour un tableau associant fièvre, polyadénopathies et hépatosplénomégalie. La NFS objective une anémie microcytaire inflammatoire sans autre cytopénie, avec lymphocytes réactionnels. L’hypothèse d’un syndrome mononucléosique est faite et G. rentre chez lui sous traitement symptomatique. 10 jours plus tard, il consulte de nouveau pour douleurs osseuses, en particulier de la jambe droite, et impossibilité à la marche transitoires. La NFS est inchangée, l’examen clinique retrouve uniquement les adénopathies cervicales et inguinales. Une échographie de la jambe droite est réalisée retrouvant une plage hypoéchogène du tiers fémoral avec rupture corticale en regard. Une IRM confirme alors la lésion fémorale et montre une infiltration diffuse de la moelle osseuse (os iliaque, fémurs, tibias) soulevant plusieurs hypothèses (sarcome d’Ewing ? ostéomyélite ? histiocytose ? neuroblastome ?). Le PET-scan retrouve des lésions hypermétaboliques intenses (fémur droit, aires ganglionnaires sus-diaphragmatiques et ostéolytiques diffuses) L’histologie de la lésion fémorale est en faveur d’une tumeur à petites cellules rondes, uniquement ERG+ et CD4+, de classement difficile. Les myélogrammes (sternal, iliaques) et le LCR sont non infiltrés.
Une biopsie-exérèse ganglionnaire cervicale complémentaire est réalisée dans un second temps et envoyée en parallèle au laboratoire d’hématologie où les appositions retrouvent une infiltration massive par des cellules d’aspect totalement indifférencié. Une lecture attentive retrouve de rares cellules atypiques, de petite taille, au noyau excentré et dense, évoquant des mégacaryocytes atypiques. L’immunophénotypage montre une prolifération CD45- avec négativité des marqueurs lymphoïdes dont le CD56 mais retrouve une expression des marqueurs CD41, CD61, CD4 (faible) CD13 CD33 CD117, évoquant un sarcome mégacaryoblastique. Le caryotype ganglionnaire est complexe avec trisomie 2, anomalie 3p et translocation t(1;22;5), variante à 3 chromosomes de la t(1;22) RBM15-MKL1. En immunohistochimie, les marquages complémentaires orientés se révèlent positifs pour le VIII et CD31.
Le diagnostic final retenu est donc celui d’un sarcome mégacaryoblastique avec t(1;22). G. est en cours d’induction selon le protocole ELAM02.
Conclusion. Les sarcomes granulocytaires (SG) sont des manifestations rares des hémopathies myéloïdes, en particulier LAM, pouvant précéder, accompagner ou être présents à la rechute de l’hémopathie. Ils surviennent en l’absence d’hémopathie avérée dans environ ¼ des cas. Les SG à différenciation mégacaryoblastique sont rarissimes (moins de 20 cas rapportés) et le plus souvent associés à des évolutions blastiques de SMP. Sur le plan thérapeutique, les SG doivent être considérés comme des LAM.
06-60
Impact pronostic du C-KIT (C117) dans les leucémies aiguës myéloblastiques : expérience de l’EHU Oran
S. Belbachir1, M. Guenna2, A. Krim3, B. Entasoltan2, N. Yafour2, S. Belaidi2, M. Brahimi2, A. Arabi2, R. Bouhass2, A. Bekadja*2
1 Service d’hématologie et de thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie ; 2 Hématologie et thérapie cellulaire, Établissement Hospitalier Universitaire 1er-Novembre, Oran, Algérie ; 3 Hématologie et thérapie cellulaire, EHU 1er-Novembre, Oran, Algérie
Introduction. Le CD117 (C-kit receptor) est un récepteur de cytokines à activité tyrosine kinase exprimé à la surface de certaines cellules, qui se lie au stem cell factor (SCF). La Spécificité de l’expression cytométrique du CD117 dans les leucémies aiguës myéloblastiques (LAM) et sa sensibilité dans le diagnostic différentiel avec les LAL sont établies mais son impact pronostic reste controversé. La transduction de signal par CD117 joue un rôle dans la survie, la prolifération et la différenciation cellulaire au cours de l’hématopoïèse. L’objectif de cette étude est d’évaluer l’impact pronostic de l’expression cytométrique du CD117 en termes de rémission complète (RC), de durée d’aplasie postchimiothérapie d’induction, de SG et de PFS et de survie postallogreffe.
Patients et méthodes. Nous avons rétrospectivement analysé les données des patients adultes atteints de LAM recrutés à l’EHU Oran sur une période de 2009 à 2017. Le diagnostic et la classification de LAM sont établis par cytologie, cytochimie, cytométrie en flux et cytogénétique. L’expression du CD117 est recherchée par cytométrie en flux. Les patients ont été répartis en deux groupes : G1 = c-kit (+) et G2 = c-kit (-). Les courbes de survie ont été établies selon la méthode de Kaplan-Meier.
Résultats. Parmi un total de 86 pts évaluables, 50 pts sont c-kit (+) (G1) et 36 pts c-kit (-) (G2).
Le taux de RC de G1 = c-kit (+) est de 71 % et de 65 % dans G2 = c-kit (-), l’expression du c-kit n’est pas prédictive du taux de réponse complète (p = 0,7). La durée d’aplasie post-chimiothérapie d’induction n’a pas été impactée par l’expression du c-kit avec une médiane de 22 jours (15-48) dans G2 = c-kit (+) et de 23 jrs (13-40) dans G2 = c-kit (-). Du point de vue évolutif, l’expression du CD117 est associée à un avantage significatif de SG (p = 0,002) avec une survie médiane de 16 mois dans G1 = c-kit (+) et de 9 mois dans G2 = c-kit (-). De même, le CD117 impact positivement la PFS (p = 0,004) avec une survie médiane de 13 mois dans G1 = c-kit (+) et de seulement 6 mois dans G2 = c-kit (-).
Parmi les pts ayant subi une allogreffe (n = 27) ou une autogreffe (n = 1), 43% sont c-kit (-) (G2) et 57 % sont c-kit (+) (G1). Leur SG est significativement supérieure dans G1 = c-kit (+) (p = 0,03) avec des médianes de 19 et 13 mois pour les c-kit (+) et c-kit (-) respectivement. Par contre, en terme de PFS, il n’a pas été retrouvé de différence significative (p = 0,06) avec des médianes de 10 et 6 mois pour les c-kit (+) et c-kit (-) respectivement.
Conclusion. Dans ce travail, la présence du CD117 au cours des LAM, a impacté positivement la survie avec une meilleure SG et PFS mais n’a pas d’impact significatif sur la réponse ou la durée d’aplasie post-chimiothérapie d’induction. Chez les pts allogreffés, le CD117, a un impact sur la SG mais pas sur la PFS. Enfin, inclure le CD117 dans le panel de routine de diagnostic des LAM doit être standardisé pour mieux déterminer son impact pronostic.
06-61
Prise en charge des patients atteints de leucémie aiguë myéloïde en France : étude nationale à partir de registres du cancer
M. Mounier1, M. Sobh2, A. Monnereau3, X. Troussard4, M. Maynadié5, M. Michallet*2
1 Registre des hémopathies malignes de côte d’or, Université de Bourgogne, Dijon ; 2 Hématologie, Centre Léon Bérard, Lyon ; 3 Registre des hémopathies malignes de la gironde, Institut Bergonié, Bordeaux ; 4 Laboratoire d’hématologie - registre régional des hémopathies malignes de Basse-Normandie, CHU de Caen, Caen ; 5 Hématologie, CHU Dijon, Dijon
Introduction. Le but principal de notre étude est d’évaluer les caractéristiques et la prise en charge de patients adultes atteints de leucémie aiguë myéloïde (LAM) entre 2006 et 2012 à partir de trois registres régionaux du cancer représentatifs de l’ensemble de la population française.
Patients et méthodes. Au total, 1 106 patients ont été évalués (446 dans le département de la Gironde, 472 en Basse-Normandie et 188 dans le département de la Côte d’Or) ; 584 (53 %) étaient des hommes ; l’âge au diagnostic était comparable entre les trois départements avec une médiane de 66 ans (18-99), 431 (39 %) avaient moins de 65 ans et 437 (39 %) avaient plus de 75 ans, 126 (11 %) avaient une LAM secondaire. L’analyse cytogénétique, hétérogène entre les trois départements [faite pour 815 (74 %) patients], a montré que 69 (8 %) patients avaient un pronostic favorable, 121 (15 %) intermédiaire et 359 (44 %) défavorable.
Résultats. Deux tiers des patients (N = 725, 66 %) ont été suivis dans des CHU, 7 % dans des centres anticancéreux et le reste des patients dans d’autres établissements de santé. Sur l’ensemble de la population, 915 (83 %) ont été traités, en majorité âgés de moins de 75 ans, et le reste des patients ont reçu des soins palliatifs. En ce qui concerne les patients de plus de 75 ans, 67 % ont été traités. Nous n’avons pas observé de différence statistique en termes de survie globale (SG) entre les hommes et les femmes (p = 0,99). La probabilité de SG à 5 ans stratifiée sur l’âge est de 50 % chez les patients de moins de 60 ans, de 20 % entre 60 et 65 ans, de 16 % entre 66 et 75 ans et de 2 % au-delà de 75 ans. L’information concernant l’inclusion dans un essai clinique était disponible chez 458 patients (50 %) : 174 (19 %) ont été inclus et 284 (31 %) n’ont pas été, avec un avantage hautement significatif en termes de SG pour les patients inclus dans un essai clinique quel que soit l’âge (p < 0,001). Les patients traités dans les CHU et les centres anticancéreux ont une survie significativement meilleure que les patients traités dans les autres établissements de santé (p < 0,001). Les résultats en termes de survie se sont nettement améliorés chez les patients âgés de moins de 65 ans au cours du temps quand on compare les périodes 2010-2012 et 20.
06-2009 (p = 0,04) mais ce n’est pas le cas des patients âgés de plus de 65 ans (p = 0,41). De plus, les patients ayant reçu une allogreffe de cellules souches hématopoïétiques (allo) ont une meilleure SG, p = 0,01. L’analyse multivariée sur la SG a montré un impact significatif de l’âge < 65 ans (HR = 0,45, IC95% : 0,38-0,54, p < 0,001), de la cytogénétique favorable et défavorable (HR = 0,26, IC95% : 0,15-0,45, p < 0,001 ; HR = 1,61, IC95% : 1,32-1,97, p < 0,001) et du fait d’avoir reçu une allogreffe (HR = 0,49, IC95% : 0,38-0,64, p < 0,001).
Conclusion. Cette analyse a permis de mettre en évidence un certain nombre de disparités quant à la prise en charge des patients porteurs d’une LAM en France. Nous avons observé l’absence d’amélioration au cours du temps chez les sujets âgés, qui met en évidence la nécessité de progresser dans cette maladie et particulièrement dans cette tranche d’âge. Une évaluation est en cours en collaboration avec la SFGM-TC pour vérifier si les patients qui répondaient aux critères de greffe ont été réellement greffés et de comparer ainsi les résultats observés en fonction de la prise en charge qui a été effectuée.
06-62
Diminution de la morbimortalité précoce des leucémies aiguës myéloïdes hyperleucocytaires par une prise en charge initiale en réanimation médicale
N. Mottal*1, N. Issa2, PY. Dumas1, F. Camou2, FX. Gros1, G. Mourissoux2, M. Sauvezie1, O. Guisset2, N. Milpied1, A. Pigneux1, T. Leguay1
1 Hématologie clinique et thérapie cellulaire, CHU Bordeaux, Bordeaux ; 2 Réanimation médicale, Hôpital Saint-André - Groupe hospitalier Saint-André - CHU de Bordeaux, Bordeaux
Introduction. Les patients atteints de leucémie aiguë myéloïde (LAM) sont à haut risque de complication initiale sévère et de décès, particulièrement lorsqu’ils sont hyperleucocytaires (HL). Une collaboration étroite entre les services d’hématologie clinique et de réanimation médicale est essentielle. Lorsqu’un transfert est nécessaire, tout retard à l’admission en Réanimation est corrélé à un pronostic plus péjoratif. Le but de cette étude était de démontrer l’intérêt d’une prise en charge thérapeutique précoce en Réanimation.
Patients et méthodes. Tous les patients de plus de 18 ans avec une LAM-HL (leucocytes > 50 G/L) au diagnostic et ayant reçu une chimiothérapie intensive d’induction dans notre centre entre 2008 et 2016 ont été analysés. Depuis 2012, les patients présentant une LAM-HL étaient pris en charge en Réanimation afin de débuter la chimiothérapie d’induction. Ils étaient ensuite transférés en Hématologie pour la suite de la prise en charge. Ils étaient parfois réhospitalisés en réanimation en cas de complications liées à l’aplasie. Ces patients appartiennent au groupe « Réanimation primaire ». Les patients ayant reçu le premier jour (J1) de chimiothérapie dans le service d’Hématologie ont été divisés en deux groupes en fonction de la nécessité d’un transfert en Réanimation durant l’induction (« Réanimation secondaire ») ou non (« Pas de réanimation »).
Résultats. Cent cinquante-quatre patients au diagnostic ont été inclus dans l’analyse : 77 dans le groupe « Pas de réanimation », 18 dans le groupe « Réanimation secondaire » et 59 dans le groupe « Réanimation primaire ». Les groupes « Réanimation primaire » et « Réanimation secondaire » présentaient des caractéristiques comparables, en dehors du taux de leucocytes médian au diagnostic, plus élevé dans le groupe « Réanimation primaire » (140 vs 68 G/L, p = 0,038). La prise en charge initiale en réanimation était associée à une diminution du recours à la ventilation mécanique (VM) (18,6 % vs 44,4 %, p = 0,027) et aux amines (11,9 % vs 33,3 %, p = 0,035). Par ailleurs, on observait une réduction de la durée de la VM (en moyenne 0,24 vs 3,7 jours, p = 0,033) et de la durée moyenne de séjour en réanimation (4,4 vs 8,7 jours, p = 0,81). Enfin, était observée une réduction de la mortalité précoce à J7 dans le groupe « Réanimation primaire » par rapport au groupe « Réanimation secondaire » (10,2 % vs 16,6 %), ainsi que la mortalité à J30 (16,9 % vs 27,8 %) mais non significative.
En comparant les groupes « Réanimation secondaire » et « Pas de réanimation », les facteurs de risque de transfert secondaire en réanimation médicale étaient la présence d’une leucostase, observée à 33 % vs 6 %, (p = 0,001) respectivement, et d’une CIVD biologique, à 38,9 % vs 11,7 % (p = 0,006).
Dans l’ensemble de la cohorte, les facteurs de risques significatifs de mortalité à J30 étaient un état général altéré (Statut OMS), un sous-type de LAM4 ou 5 selon la classification FAB, un taux de leucocyte élevé au diagnostic et une CIVD clinique.
Conclusion. La stratégie d’une prise en charge initiale en Réanimation médicale pour les LAM HL est bénéfique en termes de morbimortalité à court terme, en raccourcissant considérablement le délai de prise en charge des complications liées à l’hémopathie. Les patients atteints de LAM 4 ou 5, où présentant une CIVD ou une leucostase semblent être les plus bénéficiaires de cette stratégie.
06-63
Profil cytogénétique et immunophénotypique des leucémies aiguës lymphoblastiques de l’enfant
H. Wafik*1, S. S. Cherkaoui2, H. Nezha3, N. Khoubila4, M. Abdellah1, Q. Asmaa1
1 hématologie et oncologie pédiatrique, Hôpital 20-Août, Casablanca, Maroc ; 2 Hématologie et oncologie pédiatrique Casablanca, hématologie et oncologie pédiatrique, Casablanca, Maroc ; 3 Laboratoire de bilogie HDA, laboratoire de biologie HDA, Casablanca, Maroc ; 4 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc
Introduction. Actuellement, plus de 80 % des enfants atteints de leucémie aiguë lymphoblastique (LAL) et traités par les protocoles thérapeutiques adaptés guérissent de leur maladie. Les nouvelles données concernent essentiellement les nouveaux facteurs pronostiques cytogénétiques et moléculaires. L’objectif de notre travail est de déterminer la fréquence des leucémies lymphoblastiques B et T et des anomalies cytogénétiques dans les LAL dans une population marocaine.
Patients et méthodes. Il s’agit d’une étude descriptive qui a inclus les patients âgés de moins de 20 ans atteints de LAL suivis au service d’Hématologie et Oncologie Pédiatrique à l’hôpital 20-Août durant la période de janvier 2006 à décembre 2016. L’analyse a porté sur les données cliniques, biologiques et les résultats thérapeutiques. Le diagnostic de LAL est basé sur les critères morphologiques, cytochimiques et immunophénotypiques de la classification FAB et EGIL révisés par l’OMS. Les anomalies génétiques sont recherchées par les techniques de cytogénétiques standards.
Résultats. Durant la période d’étude, 395 cas de LAL ont été diagnostiqués. L’âge médian était de 11 ans, avec un Sex-ratio M/F de 1,8. L’étude immunophénotypique a été réalisée dans 353 des cas. Les phénotypes observés étaient B dans 221 (56 %) et T dans 174 (44 %) des cas. Le caryotype était réalisé chez 277 (70 %) patients, il était normal dans 97 (35 %) cas et retrouvait des anomalies cytogénétiques dans 121 (43 %) cas. Il s’agissait d’anomalie de nombre dans 67 (55 %) cas et de structure dans 54 (45 %) cas. Un échec de culture a été noté dans 59 (21 %) cas. L’hyperdiploïde était l’anomalie cytogénétique la plus fréquente 51 (18 %) associée au taux le plus élevé de rémission complète et le plus faible de rechute.
Conclusion. Ce travail a montré une fréquence élevée du phénotype T pouvant être expliquée par le biais du recrutement et les tranches d’âge incluses. Les caractéristiques cytogénétiques étaient similaires à celles rapportées dans d’autres pays. Cependant, un grand nombre de patients ont besoin d’être étudiés en plus en cytogénétique moléculaire afin de mieux détecter les anomalies cryptiques et de mieux stratifier les facteurs pronostiques et adapter les schémas thérapeutiques.
06-64
Caractéristiques des leucémies aiguës myéloblastiques à propos de 135 cas : expérience du service d’hématologie de l’hôpital central de l’armée (Algérie)
F. Talbi*, L. Bouteldja, A. Ghassoul, M. Djilali, K. Belateche, M. Rahali, R. Abbadi, S. Menouer, S. Belakehal, FZ. Ardjoun, K. Djouadi
hématologie, hôpital Central De l’Armée DR Mohammed Seghir Nekkache, Alger, Algérie
Introduction. Nous avons réalisé une étude rétrospective monocentrique sur une cohorte de patients suivis dans notre service pour LAM sur une période de 19 ans.
Nous avons analysé les différentes caractéristiques cliniques, biologiques et évolutives des patients et nous avons recherché une corrélation entre ses variables.
Résultats. n = 136 patients entre 2005 et 2018. n = 85 hommes/51 femmes, sex-ratio = 1,66, âge moyen = 53 ± 19 ans (18-91), n = 40 patients sont âgés de 65 ans ou plus (29 %), le diagnostic est posé après un délai moyen de 28 jours (1-45). n = 29 patients ont présenté des douleurs osseuses au diagnostic (21 %), un syndrome anémique : n = 130 (96%), un syndrome hémorragique : n = 74 (54%), un syndrome infectieux : n = 58 (42%), un syndrome tumoral (en dehors d’une splénomégalie) : n = 26 (19%), une splénomégalie : n = 29 (21%), ce paramètre est corrélé au type cytologique (p = 0,000 (s)). LAM secondaire : n = 4 cas (3 %) avec une prédominance (dans ces types) des formes sans maturation : p = 0,000 (s).
Le taux moyen de GB = 41 126 ± 79 238 elts/mm3 (300-707 900), le taux d’Hb (moy) = 7,7 ± 2,7 g/dL (2-14), le taux de plq (moy) = 57 665 ± 53 683 elts/mm3 (1 000-315 000), le pourcentage de blastes circulants = 45 ± 31 (0-100) : il est corrélé au type cytologique, p = 0,000 (s).
Selon la classification FAB : LAM0 : n = 8 (6%), LAM1 : n = 14 (10%), LAM2 : n = 41 (30%), LAM3 : n = 16 (12%), LAM4 : n = 28 (21%) dont n = 2 LAM4eo, LAM5 : n = 10 (7%), LAM6 : n = 8 (6%), LAM7 : n = 1 (0,7%), LA biphénotypique : n = 1 (0,7%), LAM inclassable : n = 9 (7%).
Nous avons retrouvé une corrélation positive entre le type de LAM est l’âge < vs ≥ 65 ans, p = 0,002 (les LAM2 sont plus fréquentes chez les patients < 65 ans) mais pas de corrélation entre ce paramètre et le sexe : p = 0,92 (ns).
N = 45 patients présentaient un taux de LDH > 40 UI/L (33 %), une complication métabolique dans : n = 46 cas (elle est corrélée au type (LAM2+++): p = 0,04 (s), le taux moyen d’acide urique = 55,2 ± 31,43 mg/L (12-162).
Traitement symptomatique : support transfusionnel pendant la phase d’induction : n = 8 ± 7,35 CGPF (0-44), n = 21,49 ± 24,56 CSP (0-147), n = 3 ± 4,09 CUP (0-26). Une forte corrélation avec le traitement reçu (3+7 vs autre) : p = 0,008 (s) pour les concentrés érythrocytaires, p = 0,002 pour les concentrés plaquettaires. n = 92 patients (68 %) ont bénéficié d’un traitement spécifique, avec un protocole 3+7 dans 73 cas (54 %). n = 35 rémissions complètes obtenues en fin d’induction (26 %), n = 61 échecs (45 %), n = 111 décès (81 %). La survie globale à 24 mois = 20 %, dans un modèle multivarié : on ne retrouve pas d’influence du sexe (p = 0,21), du délai diagnostique (p = 0,12), de l’âge (p = 0,24), du syndrome tumoral (p = 0,21), de l’hyperleucocytose > 50 000 elts/mm3 (p = 0,21), des complications métaboliques (p = 0,12). Le risque de décès est corrélé au type cytologique : p = 0,03 (S), le pourcentage le plus élevé concerne dans notre étude les LAM1.
Conclusion. La leucémie aiguë myéloblastique est une affection grave, nécessitant une prise en charge rapide, le pronostic est amélioré par les thérapies innovantes et l’accès à la greffe de moelle (si donneur compatible).
06-65
Aspects évolutifs et pronostiques des leucémies aiguës myéloïdes à core binding factor
N. Rekab*, S. Taoussi, F. Lamraoui, S. Oukid, KM. Benlabiod, Y. Bouchakor Moussa, H. Brahimi, MT. Abad, M. Bradai
Hématologie, CAC, faculté de médecine, laboratoire de recherche hémopathies malignes, hémoglobinopathies, Blida, Algérie
Introduction. Les leucémies aiguës myéloblastiques à core binding factor (CBF) regroupent les LAM avec t(8;21) et inv(16)(p13;q22) ou t(16;16)(p13q22), retrouvées respectivement chez 5 à 10 % et 10 % des LAM et sont généralement d’un pronostic favorable.
Notre objectif est de présenter les caractéristiques évolutives et pronostiques des LAM CBF chez les patients (pts) âgés de moins de 70 ans.
Patients et méthodes. De 2009 à 2017 nous avons colligé 64 cas de LAM-CBF (14 %) des LAM : t(8;21) : 6 %.
Inv(16)/t(16;16) : 8 %. Il s’agit de 30 H/34 F, d’âge moyen : 35 ans (15-68). Le diagnostic a été assuré par une étude cytogénétique par caryotype et FISH.
Traitement d’induction : protocole 3+7 : daunorubicine 45 à 90 mg/m2 J1 à J3+Cytarabine 100 mg/m2 J1 à j7. Si échec cure de rattrapage : cytarabine haute dose 3 g/m2/12 h J1, J3, J5 ± Amsacrine suivie d’une consolidation par cytarabine hautes doses : 2 à 3 cures. L’allogreffe de CSH était systématique en RC1 et depuis 2017 elle n’est réalisée qu’après la deuxième RC.
Résultats. On note une hyperleucocytose > 50 000/μL (21 pts). Type de LAM : LAM 2 : (29) dont 1 avec CD19+, 3 avec dysplasie, LAM4 : (20), LAM 4 Eos(15).
À l’étude cytogénétique, une inversion 16 (n = 32), une t(16;16)(p13q22) (n = 3), dont 15 cas étaient des LAM4 Eos, 18 LAM4 et 2 LAM2. La t(8;21) : (n = 29) chez 27 LAM2 dont 3 avec dysplasie et 2 LAM4. Patients traités : 59 (dont 2 ont reçu ARAC faible dose).
L’évaluation a intéressé les 57 pts protocolaires : non évaluables : 7 (12 %) : décès en aplasie.
RC : 45 (79 %) : chez 23 pts inv(16) et 22 pts avec t(8;21), échec : 5 (9 %).
Évaluation après deuxième cure d’induction : 4 pts : RC (2), échec : (1), non évaluables (1) : décès en aplasie.
Le devenir : greffés : 16 dont (14) en RC1 et (2) en RC2, CT exclusive : vivants en RC (18), vivants en échec (1), décès : 12 (5 en RC, 7 en rechute).
Rechutes : 12 (25 %) dont 2 postgreffe : t(8;21) 8 avec un délai moyen de rechute de 7 mois : 1 associée à un sarcome granulocytaire et 4 inv(16) avec un délai moyen de rechute de 13 mois, notés chez 50 % des cas dans les formes hyperleucocytaires.
Conclusion. Dans notre cohorte la fréquence des LAM CBF est proche de la littérature : 15 % vs 20 %.
La RC est de 78 % vs 95 % probablement liée à la mortalité en induction (12 %), on a noté 25 % de rechutes surtout dans les t(8;21). Les inv(16) sont de meilleur pronostic que les t(8;21). De meilleurs résultats passent par l’amélioration du traitement de support. Intérêt pronostique et thérapeutique de l’identification des mutations FLT3, c-Kit et de l’étude la MRD surtout en postconsolidation (MRD2) pour mieux orienter les pts pour l’allogreffe de CSH en RC1.
06-66
Caractéristiques diagnostiques et résultats thérapeutiques des leucémies aiguës lymphoblastiques de phénotype T de l’enfant et de l’adolescent dans le Sud tunisien
I. Turki1, I. Frikha*1, M. Medhaffer1, N. Louati2, I. Safra3, S. Hadijji1, I. Ben Amor1, O. Kassar1, M. Ghorbel1, M. Charfi1, F. Kallel1, H. Bellaaj1, M. Elloumi1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Hématologie biologique, Centre régional de transfusion sanguine, Sfax, Tunisie ; 3 Laboratoire hématologie, Institut Pasteur, Tunis, Tunisie
Introduction. Des particularités de la population de leucémie aiguë lymphoblastique (LAL) de l’enfant, adolescent-adulte jeune (AJA) Tunisiens sont suspectées. L’analyse restreinte des LALT permettait de mieux étudier leurs caractéristiques ainsi que leur évolution sous traitement.
Patients et méthodes. Cette étude rétrospective a colligé tous les cas de LALT (enfants âgés de 1 à 15 ans et AJA âgés de 16 à 30 ans) diagnostiqués entre janvier 2000 et décembre 2015 au service d’hématologie de Sfax et traités selon le protocole pédiatrique EORTC 58 951.
Résultats. Nous avons colligé 92 cas de LAL T soit 35 % de toutes les LAL diagnostiquées durant la période d’étude. Nous avons noté une tendance à la baisse de formes T (de 43 % des LAL durant la période 2000-2007 à 28 % durant la période 2008-2015 ; p = 0,044 et Rho = 0,125). Ces LAL de phénotype T ont concerné 55 enfants (60 %) et 37 AJA (40 %). Le sex-ratio était de 2,41. Un élargissement médiastinal était retrouvé chez 51 % des cas. Une hyperleucocytose > 100 G/L a été notée dans 39 % des cas. Le caryotype était normal chez 62 % des patients. Les anomalies cytogénétiques étaient surtout l’hyperdiploïdie > 50 (10 %), des anomalies du 11q23 (10 %), un caryotype complexe (23 %), une del(6q) (23 %) et un cas de t(9;22). Au terme de la préphase, 62 % des patients étaient corticosensibles et 38 % corticorésistants. Nous avons noté 9 décès précoces soit 10 % des cas dont 3 étaient en rémission complète (RC). Le taux de RC global était de 93 %. La stratification des patients en groupes de risque était selon les bras RM2 et RE dans respectivement 56 et 44 %. L’allogreffe de moelle osseuse a été réalisée chez 10 patients parmi 33 ayant l’indication d’allogreffe en première RC. Les décès toxiques (20 % des cas) n’étaient pas corrélés avec l’âge (p = 0,23) ni le groupe thérapeutique (p = 0,92). La rechute est survenue chez 17 cas soit 21,5 % (17 % des RM2 et 27 % des RE). Sa survenue était corrélée uniquement avec la réponse à la préphase (p = 0,006). La SG, SSE et SSM à 5 ans étaient respectivement de 55, 58 et 74 %. Le type d’anomalies cytogénétiques et la réponse à la préphase influencent significativement les différents types de survies.
Discussion. La fréquence du phénotype T dans les LAL de l’enfant et de l’adolescent dans notre série (35 %) est plus élevée que celles décrites dans la littérature (<15%). Cette fréquence élevée est aussi retrouvée par d’autres séries Tunisiennes de même que par l’étude multicentrique tunisienne faite en 2005 (37 %). Les LAL T sont caractérisées par prédominance masculine nette, des formes tumorales et prolifératives. Pour la réponse initiale au traitement, La fréquence de la corticorésistance dans notre série (38 %) est concordante avec les données de littérature (37 % dans le travail du groupe de l’EORTC rapporté par H. Cavé). Ce taux de corticorésistance est plus élevé que dans les LAL B (38 % vs 12 %). Malgré ce taux important de corticorésistance dans les LAL-T, nous n’avons pas noté plus de rechutes par rapport aux formes B (21,5 % vs 33 %). Par ailleurs, la fréquence élevée de décès toxiques (20 % vs 8 % chez les LAL-B) a influencé la SG et la SSE.
Conclusion. La fréquence élevée du phénotype T dans notre population de LAL retrouvée aussi par d’autres séries Tunisiennes est à explorer. D’autre part, l’étude des résultats des LAL-T a redressé l’impression d’avoir un taux de rechute élevé à cause de la fréquence du phénotype T.
06-67
Plus de 10 % des rechutes surviennent plus de 5 ans après la rémission dans les leucémies aiguës myéloïdes avec mutation de NPM1
S. Bertoli*1, S. Tavitian1, N. Gadaud1, F. Huguet1, E. Delabesse2, E. Berard3, C. Récher1
1 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 2 Laboratoire d’hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 3 Épidémiologie, CHU de Toulouse, Toulouse
Introduction. La majorité des rechutes de leucémies aiguës myéloïdes (LAM) intervient dans la première ou deuxième année suivant l’obtention de la rémission et peu de rechutes sont diagnostiquées après 3 ou 5 ans de suivi. Ces rechutes tardives sont peu décrites, en particulier sur le plan moléculaire.
Patients et méthodes. Nous avons répertorié dans notre base de données toutes les rechutes de LAM survenant après l’obtention d’une rémission complète (RC) en un ou deux cycles de chimiothérapie d’induction, délivrée entre 2000 et 2012. Notre analyse s’intéresse particulièrement aux rechutes tardives (RT, après 3 ans de RC) et aux rechutes très tardives (RTT, après 5 ans de RC) en comparaison aux rechutes précoces (RP, rechutes avant 3 ans de RC).
Résultats. Parmi les 636 patients ayant obtenu une RC entre 2000 et 2012, 346 rechutes cytologiques ont été diagnostiquées (54,4 %). Le délai médian à la rechute était de 0,9 année (étendue, 0,1-11,9 années ; intervalle interquartile, 0,5-1,5 années). 198 rechutes sont intervenues dans la première année faisant suite à la RC (57,2 %), 82 dans la deuxième année (23,7 %), 24 dans la troisième année (6,9 %), alors que 42 rechutes sont intervenues après la troisième année (12,1 %) et 16 après la cinquième année (4,6 %). Les caractéristiques de la LAM au diagnostic telles que l’âge, le statut de novo, le taux de globules blancs, le risque cytogénétique, les mutations de FLT3-ITD et CEBPA ou le type d’induction étaient comparables entre RP, RT, RTT. En revanche, les mutations de NPM1 étaient plus fréquentes dans les RT (46 % vs. 28 % dans les RP < 3 années, P = 0,0532), et les RTT (67 % vs. 27 % dans les rechutes < 5 années, P = 0,0070). L’allogreffe de cellules souches en première ligne était plus fréquente dans le groupe RT (24 % vs. 14 %, P = 0,0369) et dans le groupe RTT (31 % vs. 14 %, P = 0,0748). Le taux de deuxième RC (RC2) et la survie globale à partir de la rechute (OS2) étaient meilleurs dans les groupes RT et RTT par rapport au groupe RP (RC2RP : 26 %, RC2RT : 43 %, RC2RTT : 50 % ; P = 0,0154 ; OS2RP : 4,6 mois, OS2RT : 10,8 mois, OS2RTT : 11,6 mois ; P = 0,0024).
Parmi les 142 LAM avec mutation de NPM1 en RC, 67 ont rechuté (47,2 %). 39 de ces rechutes sont survenues dans la première année (58,2 % des rechutes NPM1m), 14 dans la deuxième année (20,9 %), 2 dans la troisième année (3 %), alors que 12 rechutes sont survenues après la troisième année (17,9 %), 8 après la cinquième année (11,9 %) et 3 après la huitième année (4,5 %). Chez les patients avec NPM1 sauvage, les RT et RTT étaient significativement moins fréquentes (<3 années : 91,9 % ; > 3 années : 8,1 % ; > 5 années : 2,5 % ; > 8 années : 0,6 % ; P = 0,0317, 0,0037 et 0,0783 respectivement). Les rechutes avec mutations de NPM1 initiales représentaient la moitié des RT (48 %) et deux tiers des RTT (67 %). Parmi elles, le génotype NPM1m/FLT3-sauvage était majoritaire (75 % dans les RT et 88 % dans les RTT). Le statut mutationnel de NPM1 au diagnostic n’avait pas d’impact sur le taux de RC2 ou l’OS2.
Conclusion. Nos données montrent que les rechutes tardives et très tardives ne sont pas rares chez les patients avec mutation de NPM1. Si cela est confirmé dans des séries multicentriques avec un recul suffisant, les LAM avec mutation de NPM1 devront faire l’objet d’un suivi prolongé, au-delà de cinq ans de rémission complète.
06-68
Maladie résiduelle et hématopoïèse clonale BCR-ABL1 au cours du traitement des leucémies aiguës lymphoblastiques B à chromosome Philadelphie chez l’adulte : une étude du GRAAPH-2014
R. Kim*1, Y. Chalandon2, JM. Cayuela3, P. Rousselot4, M. Passet1, E. Sylva1, X. Thomas5, V. Havelange6, P. Chevallier7, F. Huguet8, N. Boissel9, C. Berthon10, G. Stüssi11, S. Maury12, S. Chantepie13, JB. Micol14, I. Plantier15, S. Chevret16, J. Soulier1, V. Lheritier17, N. Ifrah18, H. Dombret19, E. Clappier1, GRAALL
1 Laboratoire d’hématologie, Hôpital Saint-Louis (AP-HP), Paris ; 2 Département d’hématologie, Hôpital et Université de Genève, Genève, Suisse ; 3 Laboratoire d’hématologie, Hôpital Saint-Louis, Paris ; 4 Hématologie et Oncologie, CH de Versailles, Versailles ; 5 Hématologie, Centre Hospitalier Lyon Sud, Lyon ; 6 Service d’hématologie, Cliniques Universitaires Saint-Luc, Université Catholique de Louvain, Bruxelles, Belgique ; 7 Service d’hématologie clinique, Hôtel-Dieu, Nantes ; 8 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 9 Département d’hématologie, Hôpital Saint-Louis (AP-HP), Paris ; 10 Maladie du sang, CH Régional Universitaire de Lille, Lille ; 11 Clinic of hematology, Oncology Institute of Southern Switzerland, Bellinzone, Suisse ; 12 Service d’hématologie clinique, Hôpital Henri-Mondor (AP-HP), Créteil Cedex ; 13 Service d’hématologie clinique, CHU Caen, Caen ; 14 Service d’hématologie clinique, Gustave Roussy, Villejuif ; 15 Service d’hématologie clinique, CH de Roubaix, Roubaix ; 16 Service de biostatistique et information médicale, Hôpital Saint-Louis, Paris ; 17 Service d’hématologie marcel bérard, GRAALL, Lyon ; 18 Service des maladies du sang, CHU - CHU Angers, Angers ; 19 Hématologie adulte, Hôpital Saint-Louis (AP-HP), Paris
Introduction. L’évaluation de la maladie résiduelle (MRD) des LAL-B Ph+ de l’adulte repose sur la quantification du transcrit BCR-ABL1. Cependant, il a été récemment montré que l’anomalie BCR-ABL1 n’était pas toujours restreinte aux blastes lymphoïdes, soulevant la question de la signification biologique et clinique de la MRD BCR-ABL1. Nous avons donc analysé les cinétiques de MRD d’une grande série de patients traités pour LAL Ph+ de novo, en mesurant en parallèle la fusion BCR-ABL1 et les réarrangements clonaux lymphoïdes Ig/TCR.
Patients et méthodes. L’étude a comporté 103 adultes avec LAL-B Ph+ inclus dans le protocole GRAAPH-2014. Les suivis de MRD ont été réalisés après chacun des 4 premiers cycles de traitement, sur prélèvements de moelle et sang. La quantification de la MRD Ig/TCR et du transcrit BCR-ABL1 a été effectuée de façon centralisée selon les procédures standard (EuroMRD et EAC, respectivement). Pour la quantification de BCR-ABL1 génomique, les séquences des fusions BCR-ABL1 ont été déterminées par capture des introns et séquençage nouvelle génération et mise au point de systèmes de qPCR génomique fusion-spécifique.
Résultats. Nous avons d’abord comparé les taux de BCR-ABL1 génomique avec les taux de transcrit chez 21 patients et observé une très bonne corrélation (coefficient de Pearson, r = 0,87), permettant d’écarter un biais éventuel lié à des expressions variables de BCR-ABL1. La comparaison des taux de MRD Ig/TCR et transcrit BCR-ABL1 sur 583 prélèvements de suivi issus de 103 patients a ensuite mis en évidence une mauvaise corrélation en faveur de taux de BCR-ABL1 plus élevés (r = 0,40), et ce de façon répétée chez certains patients. Nous avons donc identifié un groupe de patients avec des cinétiques de MRD dissociées, définies par des résultats de MRD Ig/TCR et BCR-ABL1 discordants (positivité/négativité ou différence ≥ 1log10) sur au moins 2 temps de suivi.
Ce groupe représentait 51 % des patients (53/103). Au diagnostic, ces patients présentaient des taux sanguins plus élevés de polynucléaires neutrophiles (4,7 vs 1,8 G/L, p = 0,001) et de monocytes (0,4 vs 0,1 G/L, p = 0,02), et des taux de blastes médullaires inférieurs (84 vs 92 %, p = 0,006) suggérant la présence d’une hyperplasie myéloïde modérée. Cependant, un syndrome tumoral n’était pas plus fréquent chez ces patients. Ce groupe était enrichi mais non majoritairement constitué de cas avec fusion M-BCR-ABL1 (42 % vs 8 %, p < 0,001).
Les patients avec cinétiques dissociées présentaient moins de réponse moléculaire majeure (RMM, BCR-ABL1 < 0,1 %) après le deuxième cycle de traitement (54 % vs 96 % sur moelle, p < 0,001 ; 48 % vs 98 % sur sang, p < 0,001) et après le quatrième cycle de traitement (80 % vs 95 % sur moelle, p = 0,002; 70% vs 98 % sur sang, p < 0,001). En revanche, leurs taux de bonne réponse évaluée sur la MRD Ig/TCR (MRD1 < 10-3 et MRD2 < 10-4) n’étaient pas différents de l’autre groupe (65 % vs 72 %, p = 0,51).
Conclusion. Cette étude met en évidence une entité de LAL-B Ph+ de novo présentant une persistance de cellules non lymphoblastiques exprimant BCR-ABL1 en rapport avec une hématopoïèse clonale. Cette entité représente environ la moitié des LAL-B Ph+ de l’adulte et comprend autant de cas avec fusions m- que M-BCR-ABL1. La signification clinique de l’hématopoïèse clonale BCR-ABL1 et le devenir à long terme de ces patients seront évalués dans le protocole GRAAPH-2014. Pour autant, ces données suggèrent que la MRD Ig/TCR sera importante pour guider les choix thérapeutiques, notamment le recours aux immunothérapies.
06-69
Challenge diagnostique des mastocytoses systémiques associées à une néoplasie hématologique : autour d’un cas
L. Philippe*1, J. Osman2, S. Marc2, L. Jaffrelot1, D. Canioni3, V. Raggueneau2, P. Rousselot4
1 Hématologie, CH de Versailles André Mignot, Le Chesnay ; 2 Laboratoire d’hématologie, CH de Versailles André Mignot, Le Chesnay ; 3 Service de cytologie et anatomie pathologiques, Hôpital Necker, Paris ; 4 Hématologie et Oncologie, CH de Versailles, Versailles
Introduction. La mastocytose systémique (SM) est une hémopathie caractérisée par une activation et/ou la prolifération clonale de mastocytes (MC) tumoraux dans au moins un organe extra-cutané selon la classification OMS 2016. Du fait de son hétérogénéité clinique et biologique, la SM est subdivisée en 5 sous-groupes. Parmi eux, la SM associée à une néoplasie hématologique (SM-AHN) (environ 1/3 des SM), combine une infiltration mastocytaire clonale et une hémopathie non mastocytaire. Dans la majorité des cas (80-90 %), il s’agit d’une hémopathie myéloïde de type leucémie aiguë myéloblastique (LAM), un syndrome myéloprolifératif et/ou myélodysplasique. Les deux hémopathies peuvent se manifester successivement ou au contraire simultanément ce qui constitue un véritable challenge diagnostique clinique et biologique. En effet, les manifestations cliniques sont principalement liées à l’atteinte médullaire tandis que sur le plan biologique, l’infiltrat médullaire de MC clonaux peut être initialement masqué par l’hémopathie non mastocytaire. Nous rapportons ici le cas d’une SM-AHN mise en évidence a posteriori d’une chimiothérapie d’induction de LAM.
Résultats. Un homme de 30 ans a été hospitalisé pour exploration de gingivorragies et altération de l’état général. L’hémogramme montrait une pancytopénie, associée à une hyperéosinophilie (3,61 × 109/L) et des blastes circulants (64 × 109/L). Le myélogramme retrouva une infiltration par 70 % de myéloblastes avec un ou deux corps d’Auer confirmant le diagnostic de LAM, et 2 % de MC dysplasiques. Les analyses moléculaires ont retrouvé deux mutations de CEBPα et la surexpression de WT1, sans transcrit BCR-ABL. Le caryotype était normal. Les examens parasitologiques réalisés étaient négatifs.
Le myélogramme à J15 d’une chimiothérapie intensive d’induction classique de type « 3+7 » à base d’anthracycline et de cytarabine, retrouve une moelle hypoplasique infiltrée par 35 % de MC dysplasiques de phénotype CD117+ CD123+ CD2- CD25-. La biopsie ostéomédullaire a identifié des ilots de plus de 15 MC cKIT+ et tryptase+, sans expression de marqueurs aberrants (CD2 et CD25 négatifs). La mutation KITD816V par PCR allèle spécifique a confirmé rétrospectivement le diagnostic de SM-AHN.
La rémission cytologique et moléculaire complète (maladie résiduelle indétectable sur la surexpression de WT1) a été obtenue en post-induction. Malgré la persistance d’une infiltration mastocytaire médullaire (1 à 3 %), en raison de l’absence de signes cliniques reliés à la SM, le traitement n’a pas été modifié. Trois cycles de chimiothérapie de consolidation ont été administrés avec persistance de la rémission complète.
Conclusion. La persistance de MC dysplasiques après une chimiothérapie intensive de LAM a conduit à la mise en évidence d’un SM-AHN. Cette observation souligne la difficulté diagnostique et l’incidence probablement sous-estimée de cette entité.
06-70
Bénéfice de l’administration de dexaméthasone en prévention des complications vitales inaugurales des leucémies aiguës myéloïdes hyperleucocytaires : analyse basée sur l’utilisation d’un score de propension
M. Cerrano1, S. Chevret2, E. Raffoux3, F. Rabian1, S. Valade4, V. Lemiale4, R. Itzykson5, N. Boissel6, H. Dombret3, E. Azoulay4, E. Lengline*7
1 Hématologie adulte, Hôpital Saint-Louis (AP-HP), Paris ; 2 Service de biostatistique et information médicale, Hôpital Saint-Louis, Paris ; 3 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 4 Réanimation médicale, Hôpital Saint-Louis, Paris ; 5 Hématologie, Hôpital Saint-Louis, Paris ; 6 Département d’hématologie, Hôpital Saint-Louis, AP-HP, Paris ; 7 Département d’hématologie, Hôpital Saint-Louis, Paris
Introduction. Une leucocytose supérieure à 50 G/L est observée au diagnostic dans 15 % des leucémies aiguës myéloïdes. 20-25 % de ces patients décèdent dans jours suivant le diagnostic en raison de complications sévères (syndrome de lyse, leucostase, hémorragies). Actuellement la meilleure stratégie de prise en charge et/ou de prévention de ces complications est débattue en l’absence d’essai randomisé. Néanmoins, l’administration en urgence de dexaméthasone a été proposée sur la base d’une étude cas contrôle de faible effectif chez des patients avec atteinte respiratoire et une LAM de groupe FAB M4/M5. L’objectif de cette étude est d’évaluer avec un meilleur niveau de preuve le bien-fondé éventuel de l’administration de dexaméthasone en prévention des complications précoces au cours des LAM hyperleucocytaires.
Patients et méthodes. Les données concernant tous les patients de plus de 18 ans admis dans notre institution entre 06/1997 et 11/2017 avec un diagnostic de LAM hyperleucocytaire (50 G/L), ont été analysées. Les LAM3 et les rechutes étaient exclues. Les critères d’évaluation étaient : le recours à une technique de suppléance vitale, l’obtention d’une rémission complète (RC) et un échec à J90 (absence de RC/rechute/décès dans les 90 jours). Des analyses pronostiques adaptées au type de critère (tests de Wilcoxon ou tests exacts de Fisher ou modèle logistique pour les échecs à J90, modèles de Cox pour la survie) ont permi de rechercher les facteurs jouant un rôle potentiel de facteur de confusion dans l’estimation de l’effet de la dexaméthasone. Un score de propension a ensuite été construit en incluant les variables pronostiques sur au moins l’un des critères de survie, rechute, échec ou défaillance d’organe dans un modèle logistique multivarié. Un appariement de chaque sujet traité par dexaméthasone à un sujet non traité sur la valeur du score de propension a été réalisé sans remise et avec un caliper de 0,10 pour diminuer les déséquilibres entre groupes de traitement sur les facteurs de confusion potentiels.
Résultats. 251 patients ont été inclus d’un âge médian de 51 IQR (34-65) ans avec une leucocytose de 120 IQR (80-188) G/L. 13 % avaient une LAM induite ou post-SMD ou SMP. 52 % avaient un syndrome de lyse, 36 % une hypoxémie, 27 % une coagulopathie. 69 % avaient un caryotype du groupe intermédiaire selon MRC, 44 % et 40 % avaient respectivement une anomalie de FLT3 et NPM1. 78 % ont reçu de l’Hydréa avant la chimiothérapie. Une suppléance d’organe était nécessaire pour 29 % dont ventilation mécanique : 22 %, dialyse : 19 %, catécholamines : 19 %. Un échec à J90 était observé chez 32 %. L’âge, la leucocytose, le pourcentage de blaste médullaire, un SLT, un PS élevé, une hypoxémie, un caryotype défavorable et un score ELN2010 non favorable étaient associés à un échec à J90. La survie globale était associée aux variables précédentes en plus de la valeur de la créatinine et FLT3-ITD. Le sexe masculin, la leucocytose, un FAB M4-M5, une créatinine élevée, une hypoxémie, un PS élevé et une coagulopathie étaient associés aux suppléances vitales. L’estimation des effets de la dexaméthasone sur la base appariée par le score de propension montre une diminution significative du risque de survenue des différents critères : Survie globale : HR = 0,59 95%CI (0,43 to 0,81) p = 0,001. Échec à J90 : HR = 0,39 95%CI (0,23 to 0,69) p = 0,02.
Conclusion. On suggère un bénéfice de la dexaméthasone sur le risque de mortalité précoce. Une confirmation prospective multicentrique pourrait être envisagée dans le futur.
06-71
Possibilité d’interruption de la prophylaxie antifongique primaire par posaconazole chez les patients suivis pour leucémie aiguë myéloïde en aplasie prolongée dans une unité protégée de conformation récente au CHU d’Angers
M. Schwarz*1, O. Corentin2, S. Thepot3, S. Francois4, P. Abgueguen5, F. Lagarce6, M. Pihet7, M. Hunault-Berger8, A. Schmidt9
1 hématologie, CHU d’Angers, Angers ; 2 Hématologie, CHU d’Angers, Angers ; 3 Maladies du sang, CHU - CHU Angers, Angers ; 4 Hématologie, CHU - CHU Angers, Angers ; 5 Service de maladies infectieuses et tropicales, CHU Angers, Angers ; 6 Pharmacie, Centre hospitalier universitaire d’Angers, Angers ; 7 Département de biologie des agents infectieux, Centre hospitalier universitaire d’Angers, Angers ; 8 Service des maladies du sang, CHU - CHU Angers, Angers ; 9 Hématologie, CHU - CHU Angers, Angers
Introduction. La gestion du risque fongique est un enjeu majeur dans la prise en charge des patients atteints de leucémie aiguë myéloïde (LAM). Le posaconazole a l’AMM en prophylaxie antifongique primaire pour les patients atteints de LAM recevant une chimiothérapie intensive. Ce traitement expose à des effets indésirables (troubles digestifs, neurologiques, cytolyse hépatique) et à des interactions médicamenteuses. De plus, son coût est important (90 €/j). Les techniques de filtration d’air sont de plus en plus performantes. Il s’agit de savoir si dans une unité avec système de filtration d’air récent, flux décroissant sur la totalité de l’unité, couloirs et zones techniques inclus, le maintien de cette prophylaxie antifongique primaire est nécessaire. Nous avons réalisé une étude au CHU d’Angers suite à notre déménagement dans un service avec flux sur l’ensemble de l’unité.
Patients et méthodes. Tous les patients suivis pour LAM recevant une induction (I) ou une consolidation (C) en secteur protégé sont inclus sur 3 ans. Le déménagement a lieu le 15 juillet 2016, nous passons d’une unité de 15 lits comportant 10 lits avec filtration sans SAS sans flux dégradé et 5 lits à flux laminaire à une unité de 20 lits avec SAS, unité filtrée en totalité avec gradient décroissant des chambres vers l’extérieur de l’unité. 3 cohortes sont définies :
– cohorte A du 1er juin 2015 au 15 juillet 2016, service des maladies du sang de 15 lits avec prophylaxie antifongique primaire par posaconazole.
– cohorte B du 15 juillet 2016 au 15 septembre 2017, nouvelle unité avec SAS et gradient de pression décroissant de 20 lits avec prophylaxie antifongique primaire par posaconazole.
– cohorte C du 15 septembre 2017 au 15 septembre 2018, nouvelle unité sans prophylaxie antifongique primaire.
L’incidence des infections fongiques est comparée par test de Fisher avec un risque alpha à 5 %.
Résultats. 84 patients ont été inclus, pour un total de 195 hospitalisations, dont 82 inductions et 113 consolidations : 19 pts (17 I/ 16 C) cohorte A, 32 pts (37 I/46 C) cohorte B et 33 pts (28 I/ 51 C) cohorte C. La durée moyenne d’aplasie est de 15,9 j (4-42) cohorte A, de 14,3 j cohorte B (3-47) et de 14,9 j (3-47) cohorte C. Dans la cohorte A, 4 aspergilloses probables sont rapportées (4/33, 12 %). Dans la cohorte B, il est observé 8 aspergilloses probables et une mucormycose prouvée (9/83, 10,8 %).
Dans la cohorte C, 6 pts ont présenté une aspergillose probable, 1 pt une aspergillose sinusienne prouvée et 1 pt une mucormycose prouvée (8/79, 10 %). 14 aspergilloses surviennent en induction, 4 en consolidation. Concernant l’incidence des infections fongiques, il n’y a pas différence significative entre les 3 cohortes (p = 0,9). Pour les 2 mucormycoses, les 2 pts ont un profil particulier avec sortie de l’unité à plusieurs reprises du fait de problèmes de compliance. Une étude pharmacoéconomique est en cours concernant le coût, les effets secondaires et les interactions médicamenteuses.
Conclusion. Dans le contexte d’une unité protégée de conformation récente, la prophylaxie antifongique primaire par posaconazole chez les patients traités pour LAM pourrait être interrompue avec un impact financier a priori non négligeable. Ces résultats encourageants nécessitent d’être confirmés sur un plus grand nombre de patients dans une étude multicentrique.
06-72
Leucémie myéloïde aiguë traitée par gemtuzumab ozogamicine : analyse rétrospective d’une cohorte de patients dans le cadre de l’autorisation temporaire d’utilisation
M. Lion*1, N. Chaumard1, L. Gilis2, AS. Michallet2, E. Nicolas-Virelizier2, A. Belhabri2, B. Favier1
1 Pharmacie, Centre Léon Bérard, Lyon ; 2 Hématologie, Centre Léon Bérard, Lyon
Introduction. Le Gemtuzumab ozogamicine (GO) est un anticorps recombinant humanisé anti-CD33 conjugué à un agent cytotoxique, la N-acétyl-gamma-calichéamicine. Cette molécule a bénéficié d’une procédure d’autorisation temporaire d’utilisation (ATU) d’octobre 2013 à juillet 2018. Dans le cadre de l’ATU de cohorte, le GO devait être prescrit en association à la daunorubicine et à la cytarabine pour le traitement des patients âgés de 50 à 70 ans atteints de leucémie myéloïde aiguë (LAM) de novo, précédemment non traitée, et avec cytogénétique favorable ou intermédiaire ou une mutation FLT3-ITD. L’objectif de notre étude est d’analyser les dossiers des patients sous GO dans le cadre d’une ATU afin d’identifier sa place dans la stratégie thérapeutique.
Matériels et méthodes. Il s’agit d’une étude monocentrique, rétrospective et observationnelle portant sur l’analyse des dossiers des patients ayant bénéficié d’une ATU de GO de 2013 à 2018 au Centre Léon Bérard. Les critères retenus étaient : âge au diagnostic, type d’ATU octroyée, indication, taux de CD33, statut cytogénétique, statut FLT3-ITD et autres anomalies moléculaires, suivi des patients postcure de GO.
Résultats. Quinze patients ont bénéficié d’un traitement par GO dans le cadre de l’ATU. Le sex-ratio (H/F) est de 2. L’âge moyen au diagnostic est de 55,5 ans (25-73 ans). Aucun patient ne répondait aux critères d’éligibilité de l’ATU de cohorte. La totalité des demandes a bénéficié d’une ATU nominative. 9/15 (60 %) patients ont reçu le GO en association à de la daunorubicine et de la cytarabine, 6 (40 %) uniquement en association à de la cytarabine. Les pourcentages de CD33 allaient de 30 à 98 %. 10/15 (66,7 %) étaient atteints de LAM de novo, 5/15 (33,3 %) de LAM secondaire. Dans 80 % des cas, le GO a été prescrit en rechute. Les 20 % restant correspondent à des prescriptions en première ligne de traitement de LAM secondaire. 7/15 (47 %) patients sont vivants. 6/7 (86 %) patients en vie ont bénéficié d’une allogreffe conduisant à 5 rémissions complètes (RC) (2 avec RC persistante à 8 mois, 1 à 3 mois, 1 à 2 mois, 1 à 6 mois) et 1 rechute moléculaire à 12 mois. Trois des patients en RC ont reçu le GO en première ligne pour une LAM secondaire à cytogénétique défavorable. Deux d’entre eux étaient porteurs de mutations de mauvais pronostic (mutations P53, EVI1). 3/7 (43 %) patients vivants étaient atteints de LAM de novo, de cytogénétique intermédiaire, en rechute, dont 2 présentant des mutations de mauvais pronostic. Seul 1/7 patient (14 %) était atteint de LAM de novo, en rechute, à cytogénétique favorable, avec anomalie moléculaire favorable (AML1-ETO). 5/8 patients décédés (62,5 %) présentaient une maladie réfractaire, 2/8 (25 %) une rechute précoce (à 2 et 3 mois) et 1/8 (12,5 %) correspond à un décès précoce de progression. Aucun des patients décédés n’a bénéficié d’une allogreffe. 2/8 (25 %) présentaient une cytogénétique défavorable et 5/8 (62,5 %) des mutations de mauvais pronostic.
Conclusion. Cette étude a permis de valider la faisabilité de cette stratégie thérapeutique dans le cadre d’une ATU nominative et montre un bénéfice pour des patients ne répondant pas aux critères de l’ATU de cohorte. Malgré des profils de patients différents, avec des facteurs de mauvais pronostic et un taux de CD33 variable, la survie de 47 % est non négligeable et met en évidence l’intérêt du GO pour le traitement de différents profils de LAM, notamment dans le cadre d’un projet de greffe.
06-73
Devenir des patients de 60 à 75 ans atteints de leucémie aiguë myéloïde secondaire en vie réelle
S. Bertoli1, S. Tavitian1, P. Bories2, I. Luquet1, T. Comont3, A. Sarry1, F. Huguet1, E. Berard4, C. Récher*1
1 Hématologie adulte, CHU de Toulouse. Institut Universitaire du Cancer de Toulouse-Oncopole, Toulouse ; 2 Oncomip, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 3 Médecine Interne et Immunopathologie, IUCT Oncopole, Toulouse ; 4 Épidémiologie, CHU de Toulouse, Toulouse
Introduction. Une récente étude de phase 3 a montré qu’une formulation liposomale de cytarabine et de daunorubicine (CPX-351) améliorait la survie globale des patients âgés de 60 à 75 ans atteints de leucémie aiguë myéloïde (LAM) secondaire. Cette étude de phase 3 représente un exemple unique de données prospectives dans ce sous-groupe rare, fournissant une base pour des comparaisons avec des données de vie réelle. Ici, nous avons évalué rétrospectivement les caractéristiques et les résultats des patients âgés de 60 à 75 ans atteints de LAM secondaire traités en routine dans notre centre.
Patients et méthodes. Nous avons sélectionné, dans notre base de données, les patients répondants aux principaux critères de l’essai CPX-351 : 60-75 ans, indice de performance ECOG 0-2, aucun traitement antérieur pour la LAM, créatinine < 176 μmol/L, bilirubine totale < 34 μM/L, LAM avec antécédents : de SMD, de LMMC ou d’exposition à un traitement cytotoxique (chimio ou radiothérapie), ou LAM de novo avec anomalies cytogénétiques de MDS ou LAM avec dysplasie multilignée.
Résultats. Sur les 218 patients répondant aux critères de l’étude CPX-351, 181 patients (83 %) ont reçu un traitement antileucémique, soit une chimiothérapie intensive (n = 121), soit un agent hypométhylant (HMA, n = 60). Par rapport aux patients traités par chimiothérapie, les patients traités par HMA étaient plus âgés, avaient moins de globules blancs, plus souvent une LAM secondaire à un syndrome myélodysplasique et un risque cytogénétique défavorable. Chez les patients traités par chimiothérapie, le taux de réponse complète était de 69 %, la survie globale médiane était de 11 mois, tandis que la survie à 3 et 5 ans était de 21 % et 17 %, respectivement. L’analyse multivariée a montré que des ATCD de chimiothérapie (HR, 2,41 ; 95 %CI : 1,48-3,92 ; P < 0,001), le risque cytogénétique défavorable (HR, 1,90 ; 95 %CI : 1,26-2,88 ; P = 0,002) et l’allogreffe évaluée en facteur dépendant du temps (HR, 0,38 ; 95 %CI : 0,16-0,89 ; P = 0,027) représentaient les facteurs pronostiques indépendants. Chez les patients traités par HMA, le taux de réponse complète était de 15 %, la survie médiane était de 11 mois, tandis que la survie à 3 et 5 ans était de 15 % et 2 %, respectivement. L’analyse multivariée a montré qu’un taux normal d’albumine (HR, 0,38 ; 95 %CI : 0,18-0,79 ; P = 0,010) et le pourcentage de blastes médullaires > 30 % (HR, 1,85 ; 95 %CI : 1,06-3,24 ; P = 0,032) représentaient les facteurs pronostiques indépendants pour la survie.
Discussion. Dans l’essai CPX-351, le bras contrôle utilisant un « 3+7 » classique (dauno 60 mg/m2 et cytarabine 100 mg/m2) a induit un taux de réponse de 33,3 % et une survie médiane de 5,95 mois. Ces résultats paraissent très inférieurs à ceux de notre étude. Cette différence peut s’expliquer par le schéma de chimiothérapie que nous utilisons (idarubicine-cytarabine et lomustine) et par des populations d’étude qui ne sont pas strictement superposables malgré des critères de sélection similaires. En particulier, le % de patients préalablement exposés aux HMA avant la chimiothérapie était nettement supérieur dans l’essai CPX-351.
Conclusion. Bien que le pronostic des sujets âgés atteints de LAM à haut risque soit très médiocre, il existe une proportion significative de patients survivants à long terme après chimiothérapie intensive ce qui n’est pas le cas avec les agents hypométhylants. Cette étude justifie le futur essai d’intergroupe ALFA/FILO comparant CPX-351, chimiothérapie intensive et azacitidine.
06-74
Leucémie aiguë myéloïde du sujet âgé : aspects épidémiologiques, clinicobiologiques et thérapeutiques
N. Sari Hassoun*1, N. Bemmoussat2, B. Benzineb3, N. Mesli4
1 CHU Tlemcen, Tlemcen, Algérie ; 2 Hématologie clinique, CHU Dr Tidjani Damerdji, Tlemcen, Algérie ; 3 Hématologie, CHU Tlemcen Faculté de médecine Tlemcen, Tlemcen, Algérie ; 4 Hématologie, CHU Dr Tidjani Damardji, Tlemcen, Algérie
Introduction. La leucémie aiguë (LA) est une pathologie rare dont l’incidence augmente avec l’âge avec un pic médian aux alentours de 65 ans au diagnostic. Les leucémies aiguës myéloïdes (LAM) représentent 80 % des LA du sujet âgé. Pour autant, la plupart des études sont menées chez des sujets jeunes et sa prise en charge chez le sujet âgé et de ses facteurs pronostics reste peu connue. L’objectif de notre étude est de décrire les caractéristiques des épidémiologiques, cliniques, biologiques et évolutifs des patients âgés de plus de 65 ans pris en charge dans notre service.
Patients et méthodes. Il s’agit d’une étude de cohorte rétrospective, monocentrique allant de janvier 2009 à janvier 2017. Avaient été inclus tous les cas de LAM ≥ 65 ans, diagnostiqués suivant les critères de l’OMS. La classification FAB avait été adoptée. Sur le plan thérapeutique, 2 groupes ont été individualisés : Induction lourde : associant Anthracycline et cytarabine, Ceux ayant relevé d’un traitement palliatif par cytarabine faible dose ou Azacitidine.
Résultats. 33 patients ont été inclus dont l’âge moyen était de 73,7 ans avec un sex-ratio H/F de 0,83. Le suivi médian était de 6,6 mois. La majorité des LAM diagnostiquées (24) étaient De Novo (72,7%). 18,1% et 6 % étaient secondaire à un syndrome myélodysplasique et aux syndromes myéloprolifératifs chroniques respectivement. Les caractéristiques des patients sur le plan clinique à l’inclusion étaient un PS ≥ 2 à 0 chez 26 patients (78,7 %). 15,1 % des patients étaient en état de sepsis. Sur le plan des comorbidités, le score de Charlson était à 0 ou 1 chez 20 patients (60,6 %) et ≥ 2 chez 13 patients (39,4 %). Sur le plan biologique, une hyperleucocytose ≥ 50 G/L est retrouvée dans 12,1 %, le taux d’Hb moyen était de 7,97 g/L et le taux de plaquette moyen était de 84 696 mm3. On y avait dénombré 11 cas de LAM2, 8 cas de LAM5, 7 cas de LAM1, 4 cas de LAM4 et 2 cas de LAM3. Le traitement initial était une induction chez 12 patients (36,3 %), 20 patients ont relevé d’une prise en charge palliative (60,6 %). 58,3 % des patients étaient en RC après l’induction et 3 patients étaient en RC après ARAC faible dose. Le taux de décès précoces selon les critères de Kantarjian (Décès < 8 semaines) était de 24,2 %. Enfin, le taux de survie à 12 mois était de 29 %.
Conclusion. Les patients âgés ont donc intérêt à bénéficier d’une induction lorsque leur état physique le permet et après une évaluation et une maîtrise des facteurs de risques. En cas d’échec, l’utilisation des nouvelles thérapies comme l’Azacitidine apparaît comme une bonne solution permettant de prolonger la survie des patients avec une toxicité limitée.
06-75
Formulation liposomale de daunorubicine + cytarabine dans la prise en charge des leucémies aiguës myéloïdes : quelles sont son efficacité et sa tolérance en vie réelle ?
P. Leclerc*1, E. Peyrilles1, E. Raffoux2, R. Rahmé3, I. Madelaine-Chambrin4, N. Jourdan4
1 Pharmacie, Hôpital Saint-Louis AP-HP, Paris ; 2 Hématologie adulte, Assistance Publique Hôpitaux de Paris, Paris ; 3 Hématologie seniors, Hôpital Saint-Louis, Assistance Publique Hôpitaux de Paris, Paris ; 4 Pharmacie, Assistance Publique Hôpitaux de Paris, Paris
Introduction. Dans le traitement des leucémies aiguës myéloïdes (LAM), une formulation liposomale synergique de daunorubicine (44 mg) et cytarabine (100 mg) (DC) a obtenu son AMM en août 2018 en première ligne des LAM de novo avec anomalies cytogénétiques, secondaires à un traitement ou à une myélodysplasie (post-SMD, post-SMD/SMP). L’objectif de cette étude est d’évaluer l’efficacité et la tolérance du DC en vie réelle.
Matériels et méthodes. Une analyse monocentrique rétrospective sur 6 mois (07/2018 à 12/2018) a été réalisée à partir du logiciel de prescription Chimio® et du dossier patient informatisé.
Résultats. 9 patients (6 H/3 F) d’âge moyen 54 ans [29 ; 74] et de statut ECOG médian à 1 [0 ; 3] ont été traités : 6 présentaient une LAM post-SMD (67 %), 2 post-SMD-SMP (22 %) et 1 de novo à caryotype complexe (11 %). 6 patients avaient une LAM de pronostic défavorable (67 %), 2 intermédiaire (22 %) et 1 favorable avec anomalie moléculaire (11 %). DC a été instauré en première ligne conformément à l’AMM pour 8 patients (92 %). Un patient a reçu 1 ligne antérieure par un agent hypométhylant : azacitidine puis allogreffe. Tous les patients ont reçu une induction à 44-100 mg/m2 J1, J3, J5, aucun n’a reçu de seconde induction.
Concernant l’efficacité, 3 patients (33 %) ont été en réponse complète (RC) et sont en projet d’allogreffe. Un patient a été en réponse partielle composite (RCi) et 1 en clairance blastique, soit au total 44 % de RC+RCi et 55 % de réponse globale (incluant RC+RCi+clairance blastique). 2/3 patients en RC après l’induction ont bénéficié d’une consolidation à 29-65 mg/m2 J1, J3. 4 patients (44 %) ont été réfractaires. 6 patients (67 %) ont reçu une seconde ligne de traitement par azacitidine. Une greffe a été réalisée chez le patient en clairance blastique après DC puis mis sous azacitidine. Un patient réfractaire est décédé des suites de la LAM.
Concernant la tolérance, la durée moyenne d’aplasie a été de 26 jours [13 ; 40]. Un patient réfractaire a eu une neutropénie persistante. Un patient a présenté une cholestase cytolytique (GGT > 4 LSN et PAL > 2 LSN) imputable au traitement avec un pic à J28. 2 patients ont majoré leurs bilans hépatiques déjà perturbés.
Conclusion. Les résultats ont été établis sur une faible cohorte. Le taux de RC est proche : 33 % vs 37,3 % dans l’étude pivot. En revanche, des différences sont à souligner avec l’étude pivot : patients plus jeunes (54 ans vs 68 ans), durée des cytopénies plus courtes : 26 vs 35 jours et un effet indésirable non décrit (cholestase cytolytique). Aussi, il est important de souligner une contrainte de préparation due à une stabilité courte (4 h) et un coût élevé (37 800 € pour une cure d’induction de DC) en comparaison à une chimiothérapie 3+7 (500 €).
06-76
Épidémiologie et résultats cliniques et thérapeutiques des patients atteints de leucémie aiguë lymphoblastique risque élevé traités au service hospitalier d’oncologie pédiatrique de Rabat
K. Zine Filali*1, A. Kili1, M. Kababri1, K. M. El1, S. Haidouri2, M. Mikdame2, K. Doghmi2, L. Hessissen1, M. Khattab1
1 Centre d’hématologie et d’oncologie pédiatrique de l’hôpital d’enfant de Rabat, Maroc, Hôpital d’enfant de Rabat, Rabat, Maroc ; 2 Hématologie clinique, Hôpital Militaire d’Instruction Mohamed V, Rabat, Maroc
Introduction. La leucémie lymphoblastique aiguë (LAL) est une hémopathie maligne caractérisée par une transformation maligne des précurseurs lymphoïdes, Elle représentait environ 75 % des cas de leucémie chez l’enfant.
L’objectif de cette étude est de collecter les données épidémiologiques et de décrire les caractéristiques cliniques et thérapeutiques des LAL hauts risques chez la population pédiatrique du service d’oncologie pédiatrique de Rabat.
Patients et méthodes. C’est une étude rétrospective qui inclut 254 cas de LAL pédiatriques haut risque, âgés de moins d’un an à 14 ans, colligés entre 2006 et 2016, admis au service d’oncologie pédiatrique de l’hôpital d’enfants de Rabat. Tous les patients étaient atteints de leucémie aiguë lymphoblastique de sous-type (B ou T), Les enfants à haut risque ont été définis comme étant âgés de 10 ans ou plus et/ou avec un nombre de globules blancs ≥ 50 000/μL au moment du diagnostic et/ou avec une atteinte du système nerveux central et/ou LAL type T, la non-réponse au corticoïde à la préphase était également considérée comme critère de mauvais pronostic. Tous les patients ont été traités par le protocole MARALL risques élevé.
Résultats. Les données cliniques sur 254 patients atteints de LAL haut risque ont été obtenues. Ils représentaient 45 % de l’ensemble des LAL. La plupart des patients (64 %) étaient des garçons et (36 %) étaient des filles, avec un Sex-ratio H/F de 1,7.
L’âge médian au moment du diagnostic était de 8,1 ans (≤ 1 an-14 ans). La plupart des patients 148 (58 %) avaient entre 1 et < 10 ans. La moitié des patients 50 % étaient originaires du Nord du Maroc. La présentation clinique au moment du diagnostic était faite chez la plupart des patients par la fièvre 62 %, 60 % avaient un syndrome tumoral, 50 % avaient un syndrome hémorragique, 22 % se sont présentés avec un élargissement médiastinal, 3 % des patients avaient une atteinte du système nerveux central au moment du diagnostic.
La plupart des patients LAL à haut risque avaient une LAL T (52 %), tandis que (48 %) avaient une LALB. 53 % de ces patients étaient hyperleucocytaires et 27 % ont présenté une corticorésistance.
Tous les patients ont été traités selon le protocole MARALL risque élevé. La réponse au traitement était basée sur la cytologie par la réalisation du myélogramme.
La rémission complète était définie par la présence de (blastes < 5 %) au myélogramme.
Sur les 245 patients, 233 patients ont obtenu une rémission complète après la phase d’induction. Durant la période de suivi (77,5 mois) 63 patients (25 %) ont rechuté.
Les sites de rechute les plus fréquents étaient la Moelle osseuse (MO) (43 patients), l’atteinte du SNC (20 patients). Le taux de survie globale (SG) pendant une moyenne de suivi de 77,5 mois était de 51 %, 44 % sont décédés et 5 % ont été perdus de vue.
Conclusion. Il s’agit d’une étude descriptive intéressant une population pédiatrique de LAL haut risque, dont les facteurs pronostics et les critères de réponse thérapeutique se sont basés sur des données cliniques, biologiques et cytologiques. Le Protocole MARALL risque élevé, est un protocole adapté au contexte marocain, qui a permis l’obtention de bons résultats de survie chez les enfants atteints d’une LAL haut risque. Le développement des techniques de biologie moléculaire dans notre Pays nous permettra de mieux identifier les patients de risque élevé, afin de leur proposer un traitement plus intensif pour améliorer leur survie.
06-77
Efficacité et tolérance du tisagenlecleucel (CTL019) dans le traitement des leucémies aiguës lymphoblastiques B de l’enfant au jeune adulte : expérience des hôpitaux Robert Debré et Saint-Louis
ME. Dourthe*1, A. Cabannes-Hamy2, K. Yakouben1, I. Rahal2, D. Chaillou1, N. Dhédin2, E. Lesprit3, J. Naudin4, J. Roupret-Serzec1, N. Parquet5, A. Brignier6, V. Guerin7, E. Lainey8, A. Caye9, H. Cavé9, E. Clappier10, S. Mathis10, S. Caillat-Zucman11, E. Azoulay12, JH. Dalle1, I. Madelaine-Chambrin13, J. Larghero14, N. Boissel2, A. Baruchel1
1 Hématologie et immunologie pédiatrique, Hôpital Robert-Debré AP-HP, Paris ; 2 Hématologie AJA, Hôpital Saint-Louis AP-HP, Paris ; 3 Établissement français du sang, Hôpital Robert Debré, paris ; 4 Réanimation pédiatrique, Hôpital Robert Debré, paris ; 5 Hématologie, Hôpital Saint-Louis (AP-HP), Paris ; 6 Service d’aphérèse, Hôpital Saint-Louis AP-HP, Paris ; 7 Immunologie biologique, Hôpital Robert Debré, paris ; 8 Service d’hématologie biologique, Hôpital Robert-Debré, Paris ; 9 Laboratoire de Génétique Moléculaire, Hôpital Robert-Debré, Paris ; 10 Laboratoire d’Hématologie, Hôpital Saint-Louis AP-HP, Paris ; 11 Laboratoire d’immunologie et d’histocompatibilité, Hôpital Saint-Louis, Paris ; 12 Réanimation médicale, Hôpital Saint-Louis, Paris ; 13 Pharmacie, Assistance Publique Hôpitaux de Paris, Paris ; 14 Service de thérapie cellulaire, hôpital saint louis, Assistance Publique Hôpitaux de Paris, Paris
Introduction. Le traitement des phases avancées de LAL-B reste un défi majeur. Le tisagenlecleucel (CTL019, lymphocytes T autologues génétiquement modifiés pour exprimer un récepteur chimérique à l’antigène CD19) a obtenu l’AMM en Europe depuis l’été 2018 pour son utilisation chez des patients de moins de 25 ans atteints de LAL B réfractaire, en deuxième rechute ou plus, ou en première rechute post-allogreffe. L’objectif de cette étude est d’évaluer la faisabilité, la tolérance et l’efficacité du CTL019 chez des patients pris en charge dans deux centres de l’Assistance-Publique-Hôpitaux de Paris et ayant eu une aphérèse en vue d’un traitement par CTL019.
Patients et méthodes. Les patients inclus dans les essais cliniques (CCTL019A2202B ELIANA et CCTL019B2001X, n = 18) ou traités dans le cadre d’ATU nominatives ou de l’ATU de cohorte (n = 11), et ayant bénéficié d’une aphérèse entre le 1er mars 2016 et le 15 octobre 2018, ont été inclus dans cette étude.
Résultats. Vingt-neuf patients, originaires de 20 centres français, de 18 ans d’âge médian (4,5 à 29 ans), ont bénéficié d’une aphérèse : 27 présentaient une rechute après un nombre médian de 3 lignes de traitement avant l’aphérèse (1-4), 2 présentaient une LAL B réfractaire en première ligne. Dix-neuf avaient reçu une allogreffe de cellules souches hématopoïétiques (68 %). Parmi les 29 patients : 3 patients étaient en attente d’injection, 4 patients sont décédés après aphérèse (3 progressions de la LAL, 1 choc septique en situation réfractaire), 1 patient a progressé. Finalement, 21 patients de 12 ans d’âge médian (4,8-29,2 ans) ont reçu l’injection de CTL019, soit une faisabilité de 81 % (21/26, 3 patients en attente). Treize patients (62 % au total, 50 % chez les moins de 18 ans, 78 % chez les plus de 18 ans) ont présenté un syndrome de relargage cytokinique (SRC) dont 9 (43 %) de grade ≥ 3 avec un délai médian de survenue de 2 jours après l’injection (0-6). Un patient est décédé de SRC à J6 de l’injection des CTL019 après la prise en charge d’une troisième rechute médullaire réfractaire. Six patients ont présenté un événement neurologique : 5 résolutifs, 1 fatal en combinaison avec le SRC. Parmi les 20 patients évaluables à 1 mois de l’injection tous étaient en rémission cytologique avec ou sans récupération hématologique complète avec une maladie résiduelle négative pour 18 d’entre eux. Au total, 5 patients ont présenté une rechute entre 2 et 10 mois après l’injection des CTL019 : CD19 positive pour 2 patients, CD19 négative pour 3 patients. Deux patients ont reçu une deuxième injection de CTL019 à 5 et 7 mois après la première pour réapparition de lymphocytes B respectivement à 3 et 4 mois postinjection de CTL019. Après un suivi médian de 6,6 mois (0,2-24,5), 18 patients sur les 21 ayant reçu l’injection de CTL019 (86 %) étaient vivants en rémission complète, 3 patients étaient décédés. À 6 mois, pour les 21 patients, la survie sans rechute est de 77,5 % (IC95% ; 50,5-90,9 %) ; la survie globale est de 95,2 % (IC95% ; 70,7-99,3 %). L’incidence cumulée de la mortalité liée au traitement à 6 mois est de 4,8 % (IC95% ; 0-65,2 %).
Conclusion. La prise en charge coordonnée des effets secondaires avec les équipes de réanimation médicale et l’utilisation de protocoles de gestion du SRC et de la neurotoxicité permettent une mortalité précoce réduite. L’expérience française du tisagenlecleucel confirme, malgré un recul encore court, des taux de réponse très encourageants, voire une possibilité de guérison, pour ces patients lourdement prétraités, en phase avancée de LAL-B.
06-78
Intérêt des inhibiteurs de la tyrosine kinase dans le traitement des leucémies aiguës lymphoblastiques de l’adulte avec chromosome Philadelphie à propos de 13 cas
I. Frikha1, Y. Fakhfakh*1, S. Hdiji1, H. Sennena2, M. Semia3, N. Benabdeljalil4, C. Kallel5, O. Kassar1, H. Bellaaj1, A. Saad2, T. Benothmen4, H. Elleuch6, M. Elloumi1, M. Chaari6
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Laboratoire cytogénétique, CHU Farhat Hached, Sousse, Tunisie ; 3 Laboratoire biologie moléculaire, Institut Pasteur, Tunis, Tunisie ; 4 Hématologie clinique, Centre National de Greffe de la Moelle Osseuse, Tunis, Tunisie ; 5 Laboratoire hématologie, CHU Habib Bourguiba, Sfax, Tunisie ; 6 Laboratoire hématologie, CHU Hédi Chaker, Sfax, Tunisie
Introduction. Depuis l’ère de la thérapie ciblée par les inhibiteurs de la tyrosine kinase (ITK), le pronostic des leucémies aiguës lymphoblastiques (LAL) de l’adulte Philadelphie positive (Ph+) est nettement amélioré. Nous rapportons à travers cette série les résultats thérapeutiques des LAL Ph+ de l’adulte traités par les ITK associés à la chimiothérapie.
Matériels et méthodes. Notre étude est rétrospective incluant les cas de LAL Ph + de novo de l’adulte diagnostiqués au service d’hématologie clinique de l’Hôpital Hédi Chaker de Sfax durant la période allant de janvier 2009 à mars 2018. L’analyse cytogénétique et la recherche du transcrit Bcr-Abl ont été réalisées pour tous les patients. Ils ont été traités selon le protocole GRAAPH 2005. L’ITK, (Imatinib), a été utilisé à la dose de 600 à 800 mg/j. L’allogreffe de moelle osseuse a été indiquée chez les patients âgés de moins de 50 ans, en rémission complète et ayant un donneur HLA compatible intrafamilial.
Résultats. Nous avons colligé 13 cas des LAL Ph+ de novo de l’adulte. L’âge médian au diagnostic était de 44 ans (20-54 ans) et le sex-ratio était de 0,62. La présence de la t(9;22) a été retrouvée dans 8 cas parmi les patients ayant un caryotype concluant, dont 5 avaient des anomalies additionnelles. Douze avaient un transcrit Bcr-Abl positif. Un patient avait un transcrit Bcr-Abl positif mais l’étude cytogénétique n’était pas concluante. Une rémission complète (RC) a été notée dans 92 % des cas. Huit patients (âge < 50 ans) étaient en indication d’allogreffe dont sept avaient un donneur HLA compatible. Parmi ces derniers, cinq patients ont eu une allogreffe (soit 64 % des patients un donneur compatible et 45 % de toute notre population) : quatre patients sont vivants en réponse moléculaire majeure après un recul moyen de 5 ans (1-7 ans). Une patiente est décédée en postgreffe par GVH aiguë. Pour les 8 patients n’ayant pas eu une allogreffe, 4 sont malheureusement décédés par rechute, les 4 autres sont vivants en réponse moléculaire majeure après un recul moyen de 4 ans (1-7 ans).
Conclusion. Avant l’ère des ITK, les LAL Ph+ de l’adulte avaient un mauvais pronostic en dehors de l’allogreffe de moelle osseuse avec des taux de RC non satisfaisant et des survies qui n’excèdent pas 40 %. Depuis l’introduction des ITK dans les protocoles de chimiothérapies, le pronostic est nettement amélioré entraînant l’obtention d’un taux de RC dépassant 80 % dans les différentes séries de la littérature et un taux de survie sans événement à 5 ans excédant 70 %, ces mêmes constatations ont été aussi retrouvées dans notre série.
06-79
La monosomie 7 dans les leucémies aiguës myéloïdes
S. Laajouri*1, M. M. Lamchahab1, W. Matrane1, N. Hda2, B. Oukkache3, S. Cherkaoui ; N. Khoubila ; M. Qachouh ; M. Rachid ; A. Madani ; A. Quessar
1 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc ; 2 Laboratoire d’analyses de biologie médicale HDA, CHU Ibn Rochd Casa, Casablanca, Maroc ; 3 Laboratoire d’hématologie, centre hospitalier universitaire Ibn Rochd, Casablanca, Maroc
Introduction. Environ 50 % à 60 % des patients atteints de leucémie myéloïde aiguë (LAM) présentent des anomalies cytogénétiques.
Les anomalies du chromosome 7, le plus souvent sous la forme d’une délétion du bras long (7q-) ou d’une monosomie, sont mises en évidence dans 8 % des LAM de l’adulte jeune (< 60 ans). Elles sont plus fréquentes chez les sujets âgés et dans les LAM secondaires et elles confèrent un pronostic défavorable.
La monosomie 7 est la monosomie autosomique la plus fréquente des monosomies dans les LAM.
L’objective de notre travail est de décrire le profil épidémiologique et évolutif des patients adultes atteints d’une LAM avec une monosomie isolée ou un CM au sein de notre unité.
Patients et méthodes. Étude rétrospective menée sur une période de 15 ans, du 1er janvier 2003 au 31 décembre 2017, de type descriptif, Les données épidémiologiques et thérapeutiques des patients ont été collectées à l’aide d’une fiche d’exploitation à partir de la base de données cytogénétiques et de dossiers médicaux de patients. Traitement : (hydroxyurée : hyperleucocytose (≥ 50 G/L), protocole AML-MA 2003, De 2003 à 2010 et le protocole AML-MA 2011: De 2011 à 2017, Traitement palliatif des patients unfit : cytarabine et mercaptopurine ou hydroxyurée).
Résultats. Au cours de notre étude, nous avons identifié 29 patients atteints de LAM avec une monosomie, L’âge médian de ces patients était de 40 ans [1-64] et le sex-ratio H/F était de 1, 9. Dix-sept (58,62 %) de nos patients étaient hyperleucocytaires au moment du diagnostic, Selon la classification FAB, le type cytologique dominant était M2 : 10 (34,48 %) des cas. Une LAM/Myélodysplasie (MDS) était retrouvé chez 5 (17,24 %) patients. La monosomie 7 était isolée chez 21 (72,41 %) patients. Sur le plan thérapeutique, 4 patients (13,79 %) ont été perdus de vue avant le traitement, 3 (10,34) sont décédés au moment du diagnostic. Le traitement palliatif était indiqué d’emblée devant l’âge avancé, l’état général du patient et la transformation du MDS en LAM chez 4 patients (13,79 %). Seulement 18 patients (62,06 %) ont été traités, une rémission complète (RC) après deux inductions a été obtenue chez 3 patients mais les 3 patients avaient rechuté. L’échec postinduction chez 12 patients, 3 patients sont décédés au cours des cycles d’induction.
Conclusion. Le pronostic de la LAM associé à une monosomie du 7 est défavorable. Les indications thérapeutiques dans ce groupe de patients doivent être bien définies et l’allogreffe des cellules souches hématopoïétiques devrait être indiquée lors de la première RC afin d’améliorer la survie de ces patients.
06-80
Les leucémies aiguës promyélocytaires : caractéristiques cliniques, biologiques et thérapeutiques. À propos de 43 cas
W. Chenbah*1, H. Regaig2, N. Bensayed3, E. Bouslama3, B. Achour3, A. Braham4, N. Braham-Jmili5, YY. Ben3, K. Abderrahim3, H. Sennana6
1 Hématologie, Hôpital Farhat Hached Sousse, Monastir, Tunisie ; 2 Hématologie, Hôpital Farhat Hached Sousse, Sousse, Tunisie ; 3 Hématologie clinique, CHU Farhat Hached, Sousse, Tunisie ; 4 Service d’hématologie CHU Farhat Hached Sousse, CHU Farhat Hached, Sousse, Tunisie ; 5 Service d’hématologie, CHU Farhat Hached, Sousse, Tunisie ; 6 Cytogénétique, génétique moléculaire et biologie de la reproduction, CHU Farhat Hached, Sousse, Tunisie
Introduction. La leucémie aiguë promyélocytaire (LAP) constitue 1 % des leucémies aiguës myéloïdes, représentant ainsi une entité particulière myéloïde par ses caractéristiques cliniques, biologiques, cytogénétiques et thérapeutiques.
Matériels et méthodes. Nous rapportons 43 patients atteints de la LAP diagnostiqués au Laboratoire d’Hématologie et traités au Service d’Hématologie Clinique de l’Hôpital Farhat Hached de Sousse durant une période de 12 ans (janvier 2005-décembre 2017).
Résultats. L’âge moyen de nos patients est de 33,6 ans, 22 de sexe féminin et 11 de sexe masculin. Le motif de consultation est celui en rapport avec les signes d’insuffisance médullaire secondaire à l’envahissement blastique de la moelle osseuse : un syndrome anémique dans 95 % des cas, un syndrome hémorragique dans 77 % des cas et un syndrome infectieux rebelle aux antibiotiques dans 37 % des cas. L’hémogramme a montré une anémie normochrome normocytaire arégénérative dans 95 % des patients, une thrombopénie dans 93 % des cas aggravée par une coagulation intravasculaire disséminée et une leucopénie dans 33 % des cas. L’analyse cytologique de la moelle a permis le diagnostic de la LAP chez 41/43 patients avec distinction de 4 aspects morphologiques distincts : LAP classique avec souvent des corps d’Auer en fagot (33 cas), LAP microgranulaire (4 cas), LAP hyperbasophile (1 cas), LAP mixte (4 cas). La présence de t(15;17) au caryotype caractéristique de la LAP a posé le diagnostic chez les 2 autres patients dont les frottis étaient désertiques. Selon le score de SANZ, les patients sont classés en différents risques : 10 à risque élevé, 23 à risque intermédiaire et 10 à risque faible, bénéficiant tous de l’acide tout-transrétinoique et traités selon un des 2 protocoles : APL99 et AIDA. Le taux de rémission complète postinduction est de 91 % et la survie globale à 2 ans 72 %.
Conclusion. Bien que nos résultats soient satisfaisants, beaucoup d’efforts sont en cours pour améliorer le pronostic.
06-81
Les monosomies autosomiques et les caryotypes monosomiques dans les leucémies aiguës myéloïdes
S. Laajouri*1, M. M. Lamchahab1, M. Dakkoune1, MR. Massi1, N. Hda2, B. Oukkache3, S. Cherakaoui ; N. Khoubila ; M. Qachouh ; M. Rachid ; A. Madani ; A. Quessar
1 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc ; 2 Laboratoire d’analyses de biologie médicale HDA, CHU Ibn Rochd Casa, Casablanca, Maroc ; 3 Laboratoire d’hématologie, centre hospitalier universitaire Ibn Rochd, Casablanca, Maroc
Introduction. La leucémie aiguë myéloïde (LAM) représente 1 % des cancers et 80 % des leucémies aiguës de l’adulte. Le caryotype est indispensable lors d’une évaluation d’une LAM au diagnostic car les anomalies cytogénétiques détectées constituent l’un des plus puissants facteurs pronostiques indépendants de cette pathologie. Environ 60 % des LAM présentent un caryotype anormal. La survie globale des LAM à caryotype monosomique (CM) est inférieure à 5 % à 4 ans Les anomalies du chromosome 7 sont mises en évidence dans 8 % des LAM de l’adulte jeune.
L’objectif de notre travail est d’étudier les résultats thérapeutiques des patients traités pour une LAM avec une monosomie 7 dans le département d’hématologie.
Patients et méthodes. Étude descriptive rétrospective, s’étalant sur une période de 7 ans, de janvier 2011 au décembre 2017, Les critères d’inclusion (Age entre 18 ans et 60 ans, LAM de novo, une monosomie isolée sur le caryotype ou un CM). Les critères d’exclusion (Age < 18 et > 60 ans LAM3, TA LMC, LA biphénotypique, LAM secondaire, décès et PDV avant le traitement). Le CM se définit selon Breems et al. par la présence d’une monosomie (à l’exception de la perte du X ou du Y) associée à au moins une anomalie de structure ou à une autre monosomie autosomique. Les données épidémiologiques et thérapeutiques des patients ont été collectées à l’aide d’une fiche d’exploitation à partir des dossiers médicaux des malades. L’analyse statistique a été effectuée par SPSS 18,0.
Résultats. 896 patients ont été diagnostiqués LAM durant la période d’étude, seulement 21 ont été inclus dans notre étude. L’âge médian était de 38 ans (18-58), le sex-ratio H/F était de 1,1. Douze patients étaient hyperleucocytaires initialement. Selon la classification FAB, le type cytologique dominant était M2 ; observé chez 10 patients. La monosomie 7 était la monosomie la plus fréquente, elle a été retrouvée isolé chez 7 patients et dans un CM chez 2 patients, le CM était retrouvé chez 8 patients. Sur le plan thérapeutique, tous les patients ont été traités, la rémission complète (RC) post induction I était obtenue chez 6 malades et après la deuxième induction chez un seul, 5 patients sont décédés au cours de l’induction I, L’échec post les 2 inductions était observé chez 9 patients. Après un suivi médian de 6,1 mois, la RC était maintenue chez 4 patients, 2 patients avaient rechuté et un patient est décédé en fin d’induction.
Conclusion. Les caryotypes monosomiques ainsi que la monosomie du 7 sont parmi les anomalies chromosomiques qui confèrent un mauvais pronostic dans toutes les séries de LAM, les indications thérapeutiques dans ce groupe de patients doivent être bien définies et l’allogreffe des cellules souches hématopoïétiques devrait être indiquée lors de la première RC afin d’améliorer la survie de ces patients.
06-82
Étude des effets du ciblage de MCL1 par le S63845 seul ou en association avec le vénétoclax dans les leucémies aiguës myéloïdes
M. Hormi, R. Birsen, M. Belhadj Merzoug, D. Bouscary, N. Chapuis*
Inserm U1016, Institut Cochin, Paris
Introduction. Les LAM sont des pathologies de mauvais pronostic et le développement de thérapies ciblées est souhaitable afin d’améliorer la survie des patients. Les BH3 mimétiques comme l’ABT-199 (vénétoclax) constituent des thérapeutiques prometteuses et cibleraient un compartiment de cellules souches leucémiques surexprimant BCL-2. Le vénétoclax a été récemment testé chez des patients atteints de LAM avec des résultats très prometteurs notamment en association avec les agents déméthylants. Cependant, les réponses au vénétoclax restent hétérogènes et des mécanismes de résistance ont récemment été décrits dans d’autres hémopathies notamment en lien avec l’induction d’expression de la protéine antiapoptotique MCL1. Nous avons donc étudié dans les cellules de LAM les mécanismes de résistance au vénétoclax et testé les effets du ciblage de MCL1 seul et association avec le vénétoclax.
Matériels et méthodes. Les effets du ciblage de MCL1 avec le composé S63845 seul ou en association avec le vénétoclax ont été étudiés dans des lignées cellulaires et des cellules primaires de patients atteints de LAM. Les études d’apoptose ont été réalisées par un marquage Annexine V analysé par cytométrie en flux. Une lignée rendue résistante au vénétoclax (MOLM-14R) par une exposition constante à des concentrations croissantes de ce composé a également été utilisée. L’étude de l’expression des protéines antiapoptotiques BCL-2 et MCL1 a été réalisée par immunoempreinte.
Résultats. Nous avons tout d’abord observé dans la lignée rendue résistante au vénétoclax MOLM-14R une augmentation d’expression des protéines MCL1 et BCL-XL. Cette surexpression de MCL1 rend les cellules MOLM-14R hypersensibles au ciblage de MCL1 puisqu’une induction d’apoptose à 24 h est observée en présence de faibles doses de S63845 (dès 10 nM) qui sont sans effet sur la survie des cellules MOLM-14. De façon intéressante, l’augmentation de l’expression de MCL1 est induite dès 8 h d’exposition au vénétoclax (à la dose de 20 nM) dans la lignée MOLM-14. Ces résultats suggèrent donc que le ciblage de MCL1 pourrait être bénéfique en cas de résistance au vénétoclax mais également pour potentialiser les effets du ciblage de BCL-2. Nous avons ensuite observé que le S63845 induisait après 24 h d’exposition une apoptose dose dépendante des cellules MOLM-14, HL-60 et OCI-AML2 (IC50 entre 500 et 1 000 nM) mais pas des lignées K562, Kasumi, TF1 et SET2 (à la dose maximum de 1 000 nM). Cependant, la sensibilité de ces cellules au S63845 ne semble pas être liée au niveau d’expression des protéines MCL1 et BCL-2 observé par immunoempreinte. Un effet proapoptotique du S63845 a également été observé dès la dose de 50 nM dans les cellules primaires de LAM (8 patients testés). Enfin, pour vérifier si le S63845 pouvait potentialiser les effets du ciblage de BCL-2, nous avons testé les effets combinés de faibles doses de vénétoclax et de S63548 sur la viabilité des cellules leucémiques. Une synergie importante a ainsi été mise en évidence entre les deux molécules dans les lignées OCI-AML2 et MOLM-14 et sur les cellules primaires de 3 patients.
Conclusion. L’ensemble de ces résultats suggèrent donc que l’association du ciblage de BCL-2 et de MCL1 dans les LAM pourrait constituer une stratégie thérapeutique intéressante dans ces pathologies.
06-83
Profil épidémiologique et cytologique des leucémies aiguës : à propos de 164 cas colligés à l’hôpital militaire d’instruction Mohamed V
S. Lhajoui*, H. Zahid, A. Biallaten, R. El Hadef, N. Messaoudi
Laboratoire d’hématologie et d’immunohématologie, hôpital militaire d’instruction Mohamed V, Rabat, Maroc
Introduction. La leucémie aiguë (LA) est une hémopathie maligne caractérisée par une prolifération monoclonale intramédullaire de cellules hématopoïétiques anormales dont le processus de maturation est bloqué au stade de « Blaste ».
On distingue selon l’origine du précurseur impliqué deux grands types de LA : lymphoblastiques (LAL) et myéloblastiques (LAM).
L’objectif de ce travail est de décrire les caractéristiques épidémiologiques et cytologiques des LA diagnostiquées à l’HMIMV.
Patients et méthodes. Il s’agit d’une étude rétrospective descriptive, réalisée au laboratoire d’hématologie de l’hôpital militaire d’instruction Mohamed V sur une période de 8 ans allant du 1er janvier 2010 jusqu’au 30 décembre 2018; portant sur les patients chez lesquels une LA a été diagnostiquée. Les données des patients ont été collectées à partir du site informatique du laboratoire et des registres de myélogramme et du frottis sanguin.
Résultats. Cent soixante-quatre cas de LA ont été colligés sur 8 ans, une prédominance masculine a été enregistrée avec un sex-ratio (H/F) de 1,6. L’âge moyen est de 46 ans [18 mois-86 ans].
La majorité des patients sont hospitalisés au service d’hématologie clinique (77 %), suivi du service de pédiatrie (14 %) et de la médecine interne (9 %).
L’examen clinique retrouve principalement un syndrome tumoral dans 40 % des cas, une fièvre dans 32 % des cas et un syndrome hémorragique dans 28 % des cas.
L’hémogramme a décelé une hyperleucocytose dans 59 % des cas, une anémie dans 93 % des cas, une thrombopénie dans 77 % des cas, et une blastose sanguine moyenne de 35 %. La blastose médullaire moyenne a été de 64 %.
Le myélogramme a permis de noter selon les critères FAB, 101 cas (61,58 %) de LA myéloblastique (1 cas LAM 0, 13 cas LAM 1, 37 cas LAM 2,18 cas LAM 3, 17 cas LAM 4, 8 cas LAM 5, 6 cas LAM 6 et 1 cas LAM7), 50 cas (30,48 %) de LA lymphoblastique (24 cas LALB, 19 cas LALT et 7 cas LAL type Burkitt) et 13 cas (7,94 %) de LA où la cytologie et la cytochimie n’ont pas permis de déterminer l’appartenance à une lignée donnée.
La rechute a été observée chez 17 % des cas avec 8 cas des rechutes sanguines et médullaires, et 3 cas de rechutes méningées.
Conclusion. L’hémogramme avec une lecture minutieuse des frottis Sanguins et médullaires complétée par des réactions cytochimiques permettent encore le classement de la plupart des LA. Cependant, 1’étude de d’autres marqueurs cytogénétiques, immunologiques et moléculaires, est devenue nécessaire pour confirmer le diagnostic des LAL et pour identifier des LA d’aspect atypique.
06-84
Leucémies aiguës lymphoblastiques de l’enfant classées faible risque : expérience du service d’hématologie-oncologie pédiatrique de Casablanca
H. Wafik*1, N. Khoubila2, A. Madani2, Q. Asmaa1
1 hématologie et oncologie pédiatrique, Hôpital 20-Août, Casablanca, Maroc ; 2 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc
Introduction. La leucémie lymphoblastique aiguë est le cancer le plus fréquent chez l’enfant. Les protocoles actuels de traitement permettent une survie sans événement de 80 %. Ces progrès ont été obtenus grâce à une meilleure stratification de la maladie, fondée depuis les années 1990 sur les critères du NCI (National Cancer Institute), stratifiant les LAL en risque standard (RS) ou risque élevé (HR) et la réponse précoce au traitement. Les progrès récents ont permis l’identification de nouveaux événements génétiques et une meilleure définition de la maladie résiduelle.
L’objectif de ce travail est de décrire le profil épidémioclinique et évolutif, des patients atteints de LAL, classés RS.
Patients et méthodes. Cette étude de cohorte a inclus les patients suivis pour LAL au service d’Hématologie et Oncologie Pédiatrique à l’hôpital 20-Août durant la période de mai 2006 à décembre 2016. Le diagnostic de LAL est basé sur les critères morphologiques, cytochimiques et immunophénotypiques. Les anomalies génétiques sont recherchées par les techniques de cytogénétiques standards. Les enfants étaient classés RS selon les critères du NCI : Age supérieur à 1 an et inférieur à 10 ans, Leucocytose inférieur à 50 000/mm3, Absence d’atteinte neurologique initiale, phénotype B, l’absence de t(9;22), de t(4;11) ou hypoploïdie inférieur à 44 chromosomes et la bonne réponse à la corticothérapie ainsi qu’à la chimiothérapie. Les enfants sont traités selon le protocole national du traitement des LAL (MARALL).
Résultats. Parmi les 395 cas de LAL diagnostiqués durant la période d’étude, 29 (7,3 %) étaient stratifiés RS. L’âge médian était de 4 ans (1-9 ans), le sex-ratio M/F était à 0,8. La LAL était révélée par un syndrome tumoral chez 15 (51 %) patients. La médiane des globules blancs était de 16,6 G/L (2,7-40,2). Le caryotype a été réalisé chez 16 des patients (55 %) et montrait une hyperdiploïde dans 27 % des cas.
Sept patients ont rechuté (24 %) après un délai médian de quatre ans, la rechute était médullaire dans 13 % des cas, neurologique dans 6,5 % des cas.
Sept patients étaient décédés (19 %), 5 (14 %) par progression de la maladie, et deux décès toxiques, 1 décès à l’induction par choc septique, et le deuxième à l’interphase par toxicité au méthotrexate.
Le taux de rémission complète maintenue est de 58 % après un délai médian de 6 ans.
Conclusion. Le taux des LAL de RS est faible dans cette étude, vu que l’unité recrute les enfants et adolescents jusqu’à 20 ans. Le taux de survie faible incite à une meilleure stratification de la maladie et au recours aux techniques d’évaluation de la maladie résiduelle.
06-85
Leucémie myéloïde aiguë chez l’adulte : groupe défavorable
H. Wafik*1, L. Mouna2, B. Houssou3, H. Nezha4, O. Bouchra5, S. S. Cherkaoui6, N. Khoubila7, N. Khoubila7, M. Rachid8, M. Quachouh7, A. Madani7, Q. Asmaa1
1 hématologie et oncologie pédiatrique, Hôpital 20-Août, Casablanca, Maroc ; 2 Hématologie, Hôpital 20-Août, Casablanca, Maroc ; 3 Service d’hématologie et Oncologie Pédiatrique, centre hospitalier universitaire Inb Rochd, Casablanca, Maroc ; 4 Laboratoire de bilogie HDA, laboratoire de biologie HDA, Casablanca, Maroc ; 5 Laboratoire d’hématologie, CHU Ibn Rochd Casa, Casablanca, Maroc ; 6 Hématologie et oncologie pédiatrique Casablanca, hématologie et oncologie pédiatrique, Casablanca, Maroc ; 7 Service d’hématologie clinique et d’oncologie pédiatrique, Hôpital 20-Août 1953, Casablanca, Maroc ; 8 Service d’hématologie clinique et oncologie pédiatrique, centre hospitalier universitaire Inb Rochd, Casablanca, Maroc
Introduction. La leucémie myéloïde aiguë (LMA) est un groupe de maladies hématologiques, phénotypiques et génétiquement hétérogènes. Habituellement, les marqueurs cytogénétiques sont utilisés pour stratifier les patients en trois catégories de risques : favorable, intermédiaire, et défavorable. L’objectif de notre travail est de décrire le profil épidémiologique, cytogénétique et évolutif des patients suivis dans notre service pour LAM, groupe défavorable.
Patients et méthodes. Nous avons inclus les patients (âgés de 18 à 60 ans) atteints de LAM présentant un pronostic défavorable du 1er janvier 2011 au 31 décembre 2016. La classification pronostique de l’OMS pour 2008 a été adoptée, et les patients ont été traités par protocole AML-MA-2011.
Résultats. 630 patients adultes ont été diagnostiqués pour une LAM, 71 (11 %) avaient un pronostic défavorable. L’âge médian était de 38 ans (18-60 ans), le sex-ratio était de 1,18, 46 (65,7 %) patients sans profession, le statut de performance PS ≥ 2 chez 19 patients (26,7 %), l’hémoglobine médiane (g/dL) était 8 (3-10) ; la médiane des plaquettes (G/L) était de 45 (1-245), la médiane des leucocytes (G/L) était de 9,87 (0,8-220). Pour la cytologie, selon FAB, les types M1 : 9 (27,1 %). La myéloperoxydase était positive chez 40 patients (57,1 %). 62 patients (88,6 %) avaient un immunophénotypage. Selon le caryotype, 33 (46,4 %) caryotype complexe, 8 (11,2 %) monosomie 7, 4 (5,6 %) autre caryotype monosomal, 19 (26,7 %) del(11q23). Pour le traitement, 90 % (63/71) ont été traités par AML-MA-2011. Une rémission complète après deux inductions a été obtenue chez 25 (39,7 %) patients, échec postinduction chez 11 patients (30,2 %), 11 (30,2 %) des patients sont décédés au cours des cycles d’induction. L’OS à 24 mois est estimée à 34,5 %. L’EFS à 30 mois est estimée à 15, 5 %.
Discussion. Le taux de groupe défavorable dans notre série est comparable à la littérature (12,8 %), avec un taux plus élevé de sous-groupes 7,1 % de caryotype complexe, 4, 1 % de monosomie 7 et 1 % de del(11q23).
Conclusion. Le groupe défavorable de la LAM reste peu fréquent, avec prédominance du caryotype complexe. Ce groupe est lié à un taux élevé de mortalité, d’où l’intérêt de réaliser la biologie moléculaire.
06-86
Pronostic des leucémies aiguës lymphoblastiques de l’enfant en première rechute
I. Frikha*, I. Turki, M. Medhaffer, S. Hadijji, I. Ben Amor, M. Charfi, O. Kassar, M. Ghorbel, H. Bellaaj, F. Kallel, M. Elloumi
Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie
Introduction. Malgré les taux de guérison supérieurs à 80 % dans le traitement des LAL de l’enfant, 10 à 20 % de rechutes surviennent, y compris chez les patients de bon pronostic. Les résultats des traitements des rechutes restent limités avec des survies médiocres. Notre objectif est d’analyser les caractéristiques diagnostiques et l’évolution de la première rechute de nos LAL pédiatriques.
Patients et méthodes. Notre étude avait concerné les enfants âgés de moins de 16 ans, en rechute de LAL inclus dans le protocole EORTC 58 951 entre janvier 2000 et décembre 2015 et suivis au service d’hématologie de Sfax. La rechute médullaire (M) est définie par la réapparition dans le sang et/ou l’enrichissement en blastes (>20%) de la moelle osseuse après une période de rémission complète (RC). Une rechute combinée touche en plus une localisation extra-médullaire, neuroméningée (NM) ou gonadique (G). Le traitement proposé était le COOPRALL 1997 et 2007, la cytarabine-Novontrone ou le groupe haut risque du protocole EORTC 58 951.
Résultats. Parmi les 173 enfants en RC, 50 avaient rechuté (29 %). Il s’agit de 16 filles et 34 garçons. L’âge médian était de 7 ans. Vingt et un enfants (26 %) étaient âgés de 1 à 5 ans, 11 (26 %) âgé de 6 à 10 ans et 18 (35 %) âgé de 11 à 15 ans. Il s’agit d’une LAL de phénotype T dans 10 cas (22 %) et d’une LAL de phénotype B dans 40 cas (32 %). La fréquence des rechutes en fonction des groupes de risques était de 37 % dans le groupe RE, 18 % dans le groupe RM2, 33 % dans le groupe RM1 et 9 % dans le groupe RF. Le délai médian de survenue de la rechute était de 19 mois (2 à 71 mois). L’incidence cumulée de rechute à 1, 2 et 3 ans suivant la RC était respectivement de 40, 58 et 82 %. Une rechute précoce < 18 mois de la RC a été notée dans 46 % des cas et 2 rechutes sont survenues au-delà de 5 ans. Le délai de la rechute n’était pas corrélé au groupe de risque thérapeutique (p = 0,22). Il s’agit de rechute médullaire isolée dans 50 % des cas, NM isolée dans 8 % des cas et de rechutes combinées dans 23 % des cas (M+NM = 8%, M+G = 6% et M+NM+G = 6%). Les facteurs prédictifs de rechute étaient la réponse à la préphase et la RC à 1 cure de chimiothérapie (p respectif de 0,0002 et 0,018). Une chimiothérapie de rattrapage était instaurée pour seulement 33 patients (66 %). Le protocole thérapeutique utilisé était le COOPRALL 1997 dans 12 cas (35 %), COOPRALL 2007 dans 16 cas (47 %), EORTC VHR pour 4 cas (12 %) et cytarabine-Novontrone pour 1 cas (3 %). La RC2 était obtenue chez 20 patients (59 %). Parmi ces 20 patients en RC2 : 6 sont allogreffés dont 4 vivants en RC, 1 décédé par GVH et 1 rechute postallogreffe. Pour les 14 non allogreffés : 4 sont vivants en RC et 10 sont décédés par rechute de la maladie. La DFS à 3 ans dans notre série était de 34 %.
Conclusion. Le taux de rechute dans notre série est de 29 %, supérieur à celui décrit dans la littérature (10 à 15 %). Ceci est expliqué en partie par la fréquence plus élevée dans notre série du phénotype T et des formes hyperleucocytaires. Malgré les progrès thérapeutiques réalisés dans le traitement des rechutes des LAL de l’enfant, la survie sans leucémie à 5 ans est de l’ordre de 30 % dans la littérature et de 34 % dans notre série. L’amélioration des résultats des rechutes repose non seulement sur l’introduction de nouvelles armes thérapeutiques (rituximab, la Clofarabine…) mais aussi par la meilleure compréhension des mécanismes des rechutes afin d’espérer avoir un traitement ciblé.
06-87
Faisabilité et résultats de l’allogreffe de la moelle osseuse dans les leucémies aiguës lymphoblastiques non Philadelphie positive de l’adulte
Y. Fakhfakh*1, I. Frikha1, N. Benabdeljalil2, A. Lakhal2, O. Kassar1, H. Bellaaj1, M. Mdhaffar1, M. Ghorbel1, I. Ben Amor1, F. Kallel1, M. Charfi1, T. Benothmen2, M. Elloumi1, S. Hdiji1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Hématologie clinique, Centre National de Greffe de la Moelle Osseuse, Tunis, Tunisie
Introduction. L’allogreffe de moelle osseuse constitue le seul traitement curateur de certaines hémopathies malignes et en particulier les leucémies aiguës lymphoblastiques (LAL). Nous étudions dans ce travail la faisabilité et les résultats de l’allogreffe de moelle osseuse pour les LAL non Philadelphie positive de l’adulte.
Matériels et méthodes. Notre étude est rétrospective incluant les cas de LAL de l’adulte de haut risque (RE) diagnostiqués au service d’hématologie de l’Hôpital Hédi Chaker de Sfax durant la période de janvier 2004 à décembre 2015. Ces patients ont été traités selon des protocoles inspirés du GRAALL 2003 et 2005. L’indication d’allogreffe de moelle osseuse est systématique chez les patients dont l’âge < 50 ans et ayant un donneur intrafamilial compatible. Pour nos patients nous avons étudié l’indication d’allogreffe, la recherche d’un donneur HLA compatible familial, la faisabilité de l’allogreffe et enfin le résultat de celle-ci qui a été réalisée au centre national de greffe de moelle osseuse à Tunis.
Résultats. Parmi les 36 cas de LAL de l’adulte non Phi+ et traité selon les protocoles GRAALL 2003 et 2005, 28 patients étaient en rémission complète (RC) et 22 patients (79 %) étaient classés RE. Vingt patients étaient en indication d’allogreffe de moelle osseuse (2 avaient un âge > 50 ans). Parmi les 20 patients ayant eu une étude HLA, 14 patients (70 %) avaient un donneur familial HLA compatible. L’allogreffe de moelle osseuse n’a pu être réalisée que pour 9 patients : soit 64 % des patients ayant un donneur HLA compatible et 45 % des patients en RC. En effet 2 patients avaient rechuté avant allogreffe et 3 patients avaient des comorbidités. Le délai médian de réalisation de l’allogreffe était de 15 semaines. Parmi les allogreffés, 6 patients (67 %) sont vivants en RC après un recul moyen de 74 mois, un patient a rechuté à 6 mois postallogreffe et 2 patients sont décédés par GVH aiguë. Parmi les 19 patients en RC et non allogreffés, 6 patients sont vivants en RC (31 %) et 1 est décédé au cours du traitement de consolidation et 12 ont rechuté. La SSE à 5 ans des patients allogreffés et de ceux non allogreffés était respectivement de 72 % et de 21 % (p = 0,065).
Conclusion. Malgré la rentabilité de l’étude HLA intrafamiliale (70 % de donneur) dans notre étude, l’allogreffe n’a pu être réalisée que pour un faible effectif de patients (45 %), d’où la nécessité d’élargir l’indication de l’allogreffe pour les sujets de plus de 50 ans ou même proposer une mini-allogreffe. En plus on peut discuter la greffe haplo-identique et élargir les sources de donneurs sur fichiers (donneurs phéno-identiques). Deux tiers des patients allogreffés de notre série n’ont pas rechuté, résultat intéressant et comparable à la littérature.
06-88
Résultats thérapeutiques des leucémies aiguës lymphoblastiques non Philadelphie positive de l’adulte du sud Tunisien
I. Frikha1, Y. Fakhfakh*1, C. Kallel2, N. Louati3, I. Safra4, H. Sennena5, N. Benabdeljalil6, H. Bellaaj1, M. Mdhaffar1, O. Kassar1, M. Ghorbel1, I. Ben Amor1, F. Kallel1, M. Charfi1, A. Saad5, H. Mnif3, T. Benothmen6, M. Elloumi1, S. Hdiji1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Laboratoire hématologie, CHU Habib Bourguiba, Sfax, Tunisie ; 3 Centre régional de transfusion sanguine, CHU Habib Bourguiba, Sfax, Tunisie ; 4 Laboratoire hématologie, Institut Pasteur, Tunis, Tunisie ; 5 Laboratoire cytogénétique, CHU Farhat Hached, Sousse, Tunisie ; 6 Hématologie clinique, Centre national de greffe de la moelle osseuse, Tunis, Tunisie
Introduction. La LAL a reconnu des progrès thérapeutiques significatifs chez l’enfant. Cependant chez l’adulte les résultats sont encore non satisfaisants. L’adaptation des protocoles actuels aux facteurs pronostiques a permis l’amélioration des résultats thérapeutiques. L’objectif de notre travail est de décrire les caractéristiques diagnostiques et les résultats thérapeutiques de nos patients suivis pour LAL non Phi+.
Matériels et méthodes. Notre étude est rétrospective incluant les cas de LAL non Phi+ de l’adulte diagnostiqués au service d’hématologie clinique de l’Hôpital Hédi Chaker de Sfax durant la période allant de janvier 2004 à décembre 2015. Le traitement repose sur l’association de polychimiothérapie selon un protocole inspiré du GRAALL 2003-2005. L’allogreffe de moelle osseuse a été indiquée chez les patients traités selon le groupe RE, âgés de moins de 50 ans, en rémission complète et ayant un donneur HLA compatible intrafamilial.
Résultats. Nous avons colligé 38 cas de LAL non Phi+. L’âge médian au diagnostic était de 42 ans (27-56 ans) et le sex-ratio était de 1,1. Le taux médian des GB était de 8,44 G/L (0,56-230 G/L). Il s’agit d’une LAL de phénotype B dans 22 cas (58 %) et d’une LAL de phénotype T dans 16 cas (42 %). Le caryotype était normal dans 59 % des cas alors que des anomalies cytogénétiques ont été retrouvées dans 41 % des cas. Une corticosensibilité et une chimiosensibilité ont été notées dans respectivement 89 % et 83 % des cas. Un taux de rémission complète (RC) a été noté dans 93 % des cas. 14 patients ont rechuté soit 50 % des cas. 20 patients en indication d’allogreffe dont 14 avaient un donneur HLA compatible et neuf ont été allogreffés. Avec un recul médian de 92 mois le taux de SG, SSR et de SSE à 5 ans était respectivement de 36 %, 47 % et de 35 %. La SSE était meilleure pour le groupe allogreffé que pour le groupe non allogreffé avec respectivement 77 % vs 24 % (p = 0,017).
Conclusion. Les caractéristiques épidémiologiques, cliniques et biologiques de notre population sont comparables par rapport à la littérature. Cependant l’âge médian était plus élevé que la littérature, ceci est expliqué par le faite que les adolescents et les adultes jeunes sont traités par des protocoles pédiatriques. Le taux de RC et de SSE est comparable aux résultats des différentes séries. Cependant le taux de rechute, 50 % dans notre série, était largement supérieur par rapport à celui décrit de la littérature (de 29 à 39 %). L’intensification de la chimiothérapie en fonction de la maladie résiduelle, une meilleure implication de l’allogreffe et l’introduction de rituximab pourrait améliorer nos résultats.
06-89
Y a-t il une modification des caractéristiques épidémiologiques et diagnostiques des leucémies aiguës lymphoblastiques de l’enfant et de l’adolescent du Sud tunisien ?
I. Turki1, I. Frikha*1, M. Medhaffer1, I. Maaloul2, B. Maalej2, N. Louati3, M. Hssairi2, F. Safi2, S. Hadijji1, H. Sennena4, O. Kassar1, I. Ben Amor1, M. Charfi1, T. Kammoun2, A. Mahfoudh2, M. Elloumi1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Service de pédiatrie, CHU Hédi Chaker, Sfax, Tunisie ; 3 Hématologie biologique, Centre régional de transfusion sanguine, Sfax, Tunisie ; 4 Laboratoire cytogénétique, CHU Farhat Hached, Sousse, Tunisie
Introduction. La leucémie aiguë lymphoblastique (LAL) est le cancer pédiatrique le plus fréquent dans le monde et en Tunisie. Les cas de LAL de l’enfant et adolescent du Sud Tunisien sont pris en charge dans le service d’hématologie de Sfax. Notre étude a pour objectif de décrire les spécificités épidémiologiques et clinicobiologiques de notre population de LAL du sud Tunisien et dégager d’éventuelles modifications de leurs aspects.
Patients et méthodes. Nous avons procédé au recueil des données des cas de LAL colligés sur 2 périodes de 8 ans chacune : période n̊ 1 (P1) de l’année 2000 à 2007 et la période N̊2 (P2) de l’année 2008 à 2015. Sont incluses les LAL âgées de 1 à 30 ans. Nous avons analysé rétrospectivement leurs caractéristiques épidémiologiques et clinicobiologiques. La similarité d’échantillonnage a permis alors d’étudier l’hétérogénéité des 2 échantillons.
Résultats. Les caractéristiques épidémiologiques et clinicobiologiques sont détaillées dans ce tableauTotal : n = 264 P1 (n = 134) P2 (n = 130) p Médiane d’âge (ans) 11 9 0,73 Age ≤ 5 ans 35 (26 %) 48 (37 %) 0,22 5 < Age < 16 ans 33 (24 %) 39 (30 %) 0,32 Age ≥ 16 ans 10 (7 %) 23 (18 %) 0,32 Sex-ratio M/F 1,56 1,32 0,28 Présence syndrome tumoral 109 (81 %) 97 (75 %) 0,18 Phénotype B 74 (57 %) 94 (72 %) 0,04 Phénotype T 56 (43 %) 36 (28 %) Atteinte neuroméningée 10,5 % 2 % 0,007 GB médian (G/l) 21 17 0,80 GB < 10 51 (38 %) 54 (41 %) 0,56 GB > 100 27 (20 %) 26 (20 %) 0,78 Caryotype normal 65 (48 %) 59 (45 %) 0,005 Échec de pousse cellulaire 15 (11 %) 34 (26 %) Anomalies cytogénétiques 54 (40 %) 39 (30 %) Hyperdiploïdie > 50 chr 12 (9 %) 12 (9 %) - t (9,22) 2 (1,4 %) 3 (1%) Del 11q23 3 (2 %) 4 (3 %)
Conclusion. En comparant les deux périodes P1 et P2, nous constatons une population devenue plus jeune que dans la série antérieure et une diminution des formes tumorales et du phénotype T tout en gardant une fréquence plus élevée par rapport aux séries occidentales. La fréquence de l’atteinte initiale du SNC est moindre au cours de P2 que P1, chiffre devenu proche des données de littérature. Sur le plan biologique, nous avons noté moins de formes très hyperleucocytaires (>100 G/L) durant P2 que P1 mais qui est 2 fois plus fréquente que dans la littérature. Dans notre série on a constaté aussi moins d’anomalies cytogénétiques par rapport à la littérature (45 % vs > 80 %). Cette fréquence moindre a été retrouvée durant P1 et encore moins durant P2, ceci est expliqué d’une part par la fréquence élevée des caryotypes non concluants et d’autre part par la non-réalisation de la méthode FISH dans la détection des anomalies cytogénétiques.
06-90
Résultats thérapeutiques des leucémies aiguës lymphoblastiques de l’adolescent et de l’adulte jeune traités selon le protocole pédiatrique EORTC58951. Expérience du service d’hématologie clinique de l’hôpital Aziza Othmana
Y. Ben Abdennebi*1, R. Berred1, L. Aissaoui1, M. Achour1, M. Bahri1, S. Kefi1, D. Jabeur1, E. Berred1, H. Ben Neji1, M. Bchir1, R. Mansouri1, K. Kacem1, W. Borgi2, F. Ben Lakhal2, S. Fekih Salem2, R. Ben Lakhal1, B. Meddeb1
1 Hématologie clinique, Hôpital Aziza Othmana, Tunis, Tunisie ; 2 Hématologie biologique, Hôpital Aziza Othmana, Tunis, Tunisie
Introduction. Au cours de ces dernières années, il a été démontré clairement que les protocoles d’inspiration pédiatrique ont permis d’améliorer d’une façon significative les résultats thérapeutiques en terme de risque de rechute et de survie chez les adolescents et les jeunes adultes.
Patients et méthodes. Entre janvier 2006 et décembre 2015, 65 adolescents et jeunes adultes âgés de 15 ans à 27 ans ayant une LAL, ont été traités selon le protocole EORTC58951 au service d’hématologie clinique de l’hôpital Aziza Othmana. Trente-deux (49,2 %) avaient un âge ≥ 18 ans, le sex-ratio est de 3, 20 (30,8 %) avaient un taux de GB ≥ 50 G/L, 4 (6 %) avaient une atteinte du SNC. Vingt-neuf (44,6 %) avaient une LAL-T, 27 (41,5 %) avaient une LAL-B, 5 (7,7 %) avaient une LA biphénotypique et 1 (1,5 %) LA indifférenciée. L’immunophénotypage était non contributif dans 3 cas. Sur le plan cytogénétique, 25 (38,5 %) avaient un caryotype pathologique.
Résultats. Tous les patients ont reçu une induction type RM2-VHR, 11 (16,9 %) étaient corticorésistants, 8 (12,3 %) étaient chimiorésistants à J19. À J 35 de l’induction, le taux de RC était de 93,8 % (n : 61), et un taux de décès en induction à 3,1 % (n : 2). L’étude de la MRD par CMF a été réalisée chez 53 patients, 32 (49,2 %) avaient une MRS < 10-3 et 4 (6,2 %) ont gardé une MRD ≥ 10-2. Vingt-cinq (39,7 %) ont été traités selon le bras RM2, 30 (47,6 %) selon le bras VHR et 8 (12,7 %) ont été greffés en RC1. Vingt-un événements se sont produits chez nos patients : 2 décès en induction, 1 échec, 1 suicide, 3 décès toxiques en RC et 14 rechutes. Avec un suivi médian de 76 mois, la SSE, la SSR et la SG à 5 ans étaient respectivement de 68,2 %, 75,4 % et 71,3 %. Douze (19 %) ont présenté une réaction allergique à la L-asparaginase, 6 (9,5 %) ont présenté une pancréatite et 6 (9,5 %) ont présenté un accident thrombotique.
Conclusion. Nos résultats sont encourageants et sont comparables à ceux de la littérature, ces protocoles pédiatriques sont faisables chez l’adulte jeune avec une toxicité gérable.
06-91
Aspects épidémiologiques, cliniques et biologiques des leucémies aiguës lymphoblastiques de l’adulte du Sud tunisien
Y. Fakhfakh*1, I. Frikha1, C. Kallel2, F. Magdiche2, N. Louati3, I. Safra4, H. Sennena5, M. Semia6, S. Hdiji1, H. Bellaaj1, O. Kassar1, M. Mdhaffar1, M. Ghorbel1, M. Charfi1, I. Ben Amor1, F. Kallel1, H. Mnif3, A. Saad5, M. Elloumi1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Laboratoire hématologie, CHU Habib Bourguiba, Sfax, Tunisie ; 3 Centre régional de transfusion sanguine, CHU Habib Bourguiba, Sfax, Tunisie ; 4 Laboratoire hématologie, Institut Pasteur, Tunis, Tunisie ; 5 Laboratoire cytogénétique, CHU Farhat Hached, Sousse, Tunisie ; 6 Laboratoire biologie moléculaire, Institut Pasteur, Tunis, Tunisie
Introduction. La LAL représente une pathologie rare de l’adulte avec une incidence de 1 nouveau cas par 100 000 habitant/an. L’objectif de notre travail est de décrire les caractéristiques épidémiologiques, cliniques et biologiques de nos patients.
Matériels et méthodes. Notre étude est rétrospective incluant les cas de LAL de l’adulte diagnostiqués au service d’hématologie clinique de l’Hôpital Hédi Chaker de Sfax durant la période allant de janvier 2004 à décembre 2015. Une fiche a été conçue pour recueillir les données épidémiologiques, cliniques et biologiques de nos patients.
Résultats. Nous avons colligé 49 cas des LAL de l’adulte dont 38 cas de LAL non Phi+ (78%) et 11 cas de LAL phi+ (22%). L’âge médian au diagnostic était de 42 ans (20-56 ans). Sur le plan clinique, le syndrome anémique a été retrouvé chez 41 patients (84 %), le syndrome hémorragique chez 13 patients (26 %) et le syndrome infectieux chez 16 patients (33 %). Le syndrome tumoral a été noté chez 31 patients : il était à type d’adénopathies isolées dans 35,5 % des cas et d’une splénomégalie isolée dans 22,5 % des cas. L’atteinte du système nerveux central a été notée dans 5 % des cas. Le taux médian de globule blanc était de 10 G/L (extrême 0,56 G/L-385 G/L. Une hyperleucocytose > 100 G/L a été notée dans 12 % des cas. Il s’agit d’une LAL de phénotype B dans 32 cas (65 %), d’une LAL de phénotype T dans 16 cas (33 %) et d’une leucémie aiguë biphénotypique dans un cas (2 %). Le caryotype était normal dans 49 % des cas alors que des anomalies cytogénétiques ont été retrouvées dans 51 % des cas dont 6 patients avaient la t(9;22). Dix avaient un transcrit BcrAbl positif et un patient avait la t(9;22) avec un transcrit négatif.
Conclusion. Les caractéristiques épidémiologiques et cliniques de notre population sont comparables par rapport à différentes séries de la littérature. Cependant quelques différences ont été notées, à savoir l’âge médian qui est plus élevé est expliqué par le fait que les adolescents et les adultes jeunes ont été traités selon les protocoles pédiatriques. Nous avons noté aussi plus de formes B et moins d’anomalies cytogénétiques par rapport aux différentes séries.
06-92
La carbenoxolone diminue la chimiorésistance des cellules de leucémie aiguë myéloïde favorisée par la niche mésenchymateuse
F. Kouzi1, K. Zibara2, J. Bourgeais3, F. Picou1, N. Gallay3, H. Dakik1, B. Roux1, R. Hleilel4, F. Gouilleux1, F. Mazurier1, F. Rouleux-Bonnin1, E. Gyan5, J. Domenech1, M. El-Sabban4, O. Herault*1
1 CNRS ERL 7001 lnox, Faculté de Médecine de Tours, Tours ; 2 Prase, Université Libanaise, Beyrouth, Liban ; 3 Service d’hématologie biologique, CHRU de Tours, Tours ; 4 Faculté de médecine, Université Américaine de Beyrouth, Beyrouth, Liban ; 5 Service d’hématologie et thérapie cellulaire, CHRU de Tours, Tours
Introduction. La niche des leucémies aiguës myéloïde (LAM) régule la progression leucémique en favorisant la quiescence et la chimiorésistance. Les cellules stromales mésenchymateuses (CSM) protègent les cellules leucémiques de l’apoptose induite par la chimiothérapie. Les jonctions gap entre les cellules hématopoïétiques et les CSM résultent de l’assemblage de connexines (Cx), et nous avons postulé qu’elles pourraient moduler la chimiorésistance dans la niche leucémique. Nous avons donc caractérisé ces jonctions gap et étudié les effets d’un bloqueur de leur assemblage, la carbénoxolone (CBX), déjà utilisé dans le traitement de certaines maladies humaines.
Matériels et méthodes. Les cellules utilisées étaient : 8 lignées de LAM (ECACC), 39 moelles de LAM (FILOthèque LAM), les cellules CD34+ de 8 moelles normales, les CSM issues des 19 moelles de LAM et de 30 moelles normales. L’expression de 20 gènes de Cx a été déterminée par qRT-PCR. Les cellules leucémiques ont été exposées à CBX ± cytarabine (AraC), pour évaluer la viabilité et la prolifération, le cycle cellulaire (Ki67/7AAD) et l’apoptose (Annexine V/7AAD) par cytométrie en flux. Elles ont été cotraitées avec des doses croissantes de CBX et d’AraC pour réaliser des isobologrammes. La phosphorylation oxydative (OCR) et la glycolyse (ECAR) ont été étudiées avec l’analyseur Seahorse, et le profilage métabolique à haut débit a été réalisé par OmniLog. La niche a été modélisée par coculture de cellules leucémiques et de CSM pendant 48 h en présence ou non CBX (150 μM) et AraC (1 μM). L’inhibition des jonctions gap par le CBX a été vérifiée par le transfert de calcéine. L’effet in vivo du CBX a été évalué dans les souris NSG développant une leucémie OCI-AML-3.
Résultats. Alors que le profil d’expression des Cx des cellules leucémiques était semblable aux cellules CD34+ normales, les CSM de LAM présentaient une signature Cx spécifique comparativement aux CSM normales. Utilisé seul, le CBX a diminué la croissance et la viabilité et induit l’apoptose des cellules leucémique de façon dose et temps dépendante, avec une IC50 d’environ 150 μM. Il est à noter que le CBX n’a aucun effet sur la viabilité des cellules CD34+ normales, ni sur les progéniteurs clonogéniques. Les isobologrammes des lignées LAM ont objectivé 3 profils différents de chimiorésistance et un effet synergique entre CBX et AraC. Le traitement au CBX a diminué l’OCR et l’ECAR des lignées cellulaires AML quelle que soit leur résistance à l’AraC, avec une incapacité à métaboliser de nombreux oses. En coculture, le contact avec les CSM a induit une mise en quiescence et une résistance à AraC ; le cotraitement des cellules leucémiques avec CBX et AraC a inhibé l’effet protecteur du contact avec les CSM, avec donc une moindre quiescence et une forte apoptose des cellules au contact des CSM (KG1a et leucoblastes sur des CSM de moelle normales ou de moelles leucémiques). Le CBX a bloqué les jonctions gap en réduisant de 48 % le transfert de calcéine entre les cellules MSC et les cellules KG1a. Enfin, le CBX a favorisé la survie des souris OCI-AML-3 et limité l’envahissement splénique et hépatique.
Conclusion. Le ciblage des jonctions gap spécifiques de la niche leucémique est une voie thérapeutique prometteuse dans les LAM. Alors qu’il a aucun effet délétère sur les cellules CD34+ normales, le CBX présente une activité antileucémique en lien avec une extinction métabolique, synergique avec l’Ara-C, et surtout intéressante sur les cellules chimiorésistantes au contact des CSM médullaires.
06-93
Sélection d’une population clonale après daratumumab en situation de leucémie aiguë lymphoblastique B en rechute/réfractaire
P. Lebreton1, T. Cazaubiel2, N. Lechevalier2, FX. Gros*1, E. Forcade1, PY. Dumas1, N. Milpied1, A. Pigneux1, T. Leguay1
1 Hématologie clinique et thérapie cellulaire, CHU Bordeaux, Bordeaux ; 2 Laboratoire d’hématologie, CHU Bordeaux, Bordeaux
Introduction. Le pronostic des leucémies aiguës lymphoblastiques B (LAL-B) en rechute ou réfractaires (R/R) reste aujourd’hui réservé, avec une survie sans rechute à 5 ans inférieure à 25 % 1,2. Les dernières avancées thérapeutiques ciblent des antigènes exprimés par les lymphoblastes, tels que le CD22 pour l’inotuzumab-ozogamicine ou le CD19 pour le blinatumomab et plus récemment les chimeric-antigen-receptor (CAR)-T cells.
Patients et méthodes. Nous rapportons l’intérêt du ciblage d’un autre antigène, le CD38, par le daratumumab, ayant permis une sélection clonale en situation réfractaire.
Résultats. Un patient de 19 ans est hospitalisé en juin 2017 devant le diagnostic d’une LAL-B CD10+ CD19+ CD22+ CD20- CD38+ sans chromosome de Philadelphie, sans envahissement neuroméningé. Il bénéficie du traitement selon le protocole GRAALL2014 sans atteindre une maladie résiduelle n̊2 (MRD2) inférieure à 10-4 en post-consolidation. En l’absence de donneur génoïde ou phéno-identique, il recevra une allogreffe haplo-identique en novembre 2017 après traitement par blinatumomab. Une première rechute cytologique cinq mois plus tard amène à un traitement par chimiothérapie intensive de type pédiatrique selon VANDA (cytarabine, mitoxantrone, dexaméthasone, sans L-asparaginase) en association au blinatumomab suivi d’infusions de lymphocytes du donneur. Malgré l’obtention d’une deuxième rémission complète avec une MRD post-rattrapage détectable inférieure au seuil 10-4, une nouvelle rechute cytologique advient quelques jours après l’arrêt d’un troisième cycle de blinatumomab d’entretien en octobre 2018. L’immunophénotypage (IP) met alors en évidence deux populations blastiques se différenciant sur l’intensité du CD19 : une population minoritaire de blastes CD19fort CD38faible et une population majoritaire CD19faible à négative CD38fort, probablement liée au traitement par blinatumomab. L’apparition de cette seconde population CD19faible rend le patient inéligible aux protocoles de traitement par CAR-T cells. Un schéma thérapeutique basé sur du daratumumab 16 mg/kg, vindésine 4 mg et dexaméthasone 20 mg à J1, J8, J15, et J22, est alors proposé. Le myélogramme réalisé à J28 conclut à un échec cytologique avec cependant une disparition du clone CD19faible CD38fort en IP. Bien que le CD38 se soit négativé, seule la population CD19fort persiste ce qui permet au patient d’être éligible à un anti-CD19 traitement par CAR-T cells actuellement en cours.
Discussion. Les LAL-B R/R voient leur pronostic récemment amélioré par le ciblage d’antigènes leucémiques qui représente l’alternative la plus innovante à ce jour, notamment les CAR-T cells autologues ciblant le CD193. L’utilisation du daratumumab dans les LAL n’est décrite qu’au travers de cas rapportés, et est en cours d’investigation en pédiatrie (NCT03384654). Chez notre patient, il a permis une sélection clonale de la population monoclonale CD19fort nécessaire pour être éligible à un traitement par CAR-T cells anti-CD19. Le mécanisme de modulation phénotypique du lymphoblaste est un mécanisme de résistance connu avec les anticorps monoclonaux. Une analyse moléculaire ciblée sur le réarrangement IgH/TCR aux différents points de rechute permettra d’affiner les conclusions.
Conclusion. L’utilisation séquentielle d’anticorps spécifiques permet une sélection de clones résistants par la disparition de l’antigène cible. L’arrêt de l’anticorps initial et l’utilisation d’un second anticorps peuvent néanmoins permettre de retrouver le clone initial.
06-94
Résultats thérapeutiques des leucémies aiguës lymphoblastiques de l’enfant avec chromosome Philadelphie : association d’un inhibiteur de tyrosine kinase et chimiothérapie
Y. Ben Abdennebi*1, R. Berred1, L. Aissaoui1, M. Bahri1, M. Achour1, R. Mansouri1, S. Kefi1, D. Jabeur1, E. Berred1, H. Ben Neji1, M. Bchir1, K. Kacem1, W. Borgi2, F. Ben Lakhal2, S. Fekih Salem2, E. Gouider2, R. Ben Lakhal1, B. Meddeb1
1 Hématologie clinique, Hôpital Aziza Othmana, Tunis, Tunisie ; 2 Hématologie biologique, Hôpital Aziza Othmana, Tunis, Tunisie
Introduction. Le pronostic des LAL avec chromosome Philadelphie s’est nettement amélioré depuis l’avènement des inhibiteurs de la tyrosine kinase bcr-abl, ce qui constitue actuellement le traitement optimal des LAL Ph+ en association avec la chimiothérapie. Cette entité de LAL est rare chez l’enfant (3 à 5 % des LAL de l’enfant) ; nous rapportons les résultats d’une série pédiatrique traitée selon le protocole EORTC58951 et Imatinib.
Patients et méthodes. Entre janvier 2006 et décembre 2016 ; 350 LAL de l’enfant âgé de 2 ans à 25 ans, ont été diagnostiquées et traitées au service d’hématologie clinique de l’hôpital Aziza Othmana ; 22 (6,2 %) avaient une LAL-B avec chromosome Philadelphie. Cinquante-neuf pour cent des patients (n : 13) avaient un âge ≥ 10 ans, on note une prédominance masculine avec un sex-ratio à 3,4. Le taux médian des GB est de 110 G/L (3,4 G/L-995 G/L). Un seul patient a été classé faible risque (âge < 10 ans et GB < 50 G/L), 2 avaient une atteinte initiale du SNC et des anomalies cytogénétiques associées ont été notées dans 36 % des cas.
Résultats. Tous les patients ont été traités selon le protocole EORTC58951 bras haut risque avec introduction de l’Imatinib dès l’induction avec un délai médian de 11 jours (4 jours-36 jours). Quarante-cinq pour cent (n : 10) des patients étaient corticorésistants au FS de J8 et 14,2 % (n : 3) étaient chimiorésistants au J19.
Un patient est décédé par leucostase à J3 et 2 sont décédés par choc septique en induction. Le taux de RC à J35 IA est de 86,3 % (19/22) ; les 19 patients étaient en rémission cytogénétique, 12 (63,1 %) étaient en réponse moléculaire et l’étude de la MRD par CMF a été réalisée chez 16 patients ; 9 avaient une MRD < 10-4. Le typage HLA a été réalisé chez tous les patients ; 4 avaient un donneur géno-identique. Cinq patients ont bénéficié d’une allogreffe en RC1 : 3 greffes géno-identiques et 2 greffes de cordon. Les 14 autres patients ont été traités selon le protocole EORTC58951 bras haut risque et Imatinib en continu.
Avec un suivi médian de 33 mois, la SG, la SSE et la SSR à 3 ans étaient respectivement de 64,9 %, 60,3 % et 79,1 % et sont de 64 %, 37,7 % et 49,5 % à 5 ans. Deux parmi les 5 patients greffés en RC1 sont décédés en postgreffe par infection. Parmi les 14 autres patients traités par chimio et ITK, 1 a été perdu de vue, 6 ont rechuté avec un délai médian de 36 mois (17 mois-57 mois). La SSE est passée de 75,2 % à 3 ans à 37,6 % à 5 ans pour les patients traités par chimiothérapie et ITK et est de 60 % aussi bien à 3 ans et à 5 ans pour ceux qui ont été allogreffés en RC1.
Conclusion. Le pronostic des LAL avec chromosome Philadelphie s’est nettement amélioré par l’association des ITK et chimiothérapie. Cependant des rechutes tardives ont été notées chez les patients non allogreffés en RC1.
06-95
Utilisation du blinatumomab en association a la chimiothérapie standard en cytoréduction en situation de leucémies aiguës lymphoblastiques B en rechute/réfractaire : analyse rétrospective
P. Lebreton, FX. Gros*, PY. Dumas, E. Forcade, N. Milpied, A. Pigneux, T. Leguay
Hématologie clinique et thérapie cellulaire, CHU Bordeaux, Bordeaux
Introduction. À l’ère des chimiothérapies intensives, les leucémies aiguës lymphoblastiques B (LAL-B) de l’adulte en rechute ou réfractaires (R/R) ont conservé un pronostic très réservé. Le développement d’anticorps monoclonaux et du blinatumomab en monothérapie a permis une amélioration significative en termes de survie globale au prix d’une importante toxicité liée au syndrome de relargage cytokinique (CRS) et à la toxicité neurologique induite. Peu d’essais mentionnent l’association du blinatumomab à une chimiothérapie de cytoréduction de rattrapage standard qui permettrait d’améliorer la tolérance tout en augmentant la survie. Nous rapportons notre expérience clinique évaluant chimiothérapie de cytoréduction en association au blinatumomab.
Patients et méthodes. Il s’agit d’une étude rétrospective monocentrique menée de janvier 2012 à décembre 2018 sur 9 patients porteurs d’une LAL-B sans chromosome de Philadelphie (Ph-) R/R CD19+ en première rechute cytologique.
Résultats. L’âge médian était de 42 ans (18-74). Quatre étaient primoréfractaires, 3 avaient une atteinte neuroméningée et 1 une rechute post-allogreffe. Huit patients ont été traités selon le protocole d’inspiration pédiatrique VANDA (cytarabine, mitoxantrone, dexaméthasone, sans L-asparaginase) et 1 selon le protocole EWALL-backbone (vincristine, idarubicine). Le blinatumomab était engagé en médiane à J9 (J-5-J28). Le nombre médian de cycles complets de blinatumomab administrés était de 2 (1-3). La durée moyenne de neutropénie était de 15 jours (10-26). Huit patients ont développé un CRS, dont 2 (22 %) de grade ≥ 3. Un patient a présenté une complication neurologique (grade 3). Sept ont présenté une aplasie fébrile documentée dont un cas létal. Sept patients (78 %) étaient en rémission complète en sortie d’aplasie, et 4 (44 %) ont reçu une allogreffe hématopoïétique, avec rémission moléculaire majeure (RMM) actuelle à 13, 17, 29 et 74 mois. Avec un suivi médian de 8 mois, la survie sans événement à 1 an était de 74 %.
Discussion. Notre étude sur une petite cohorte de patients adultes de notre centre démontre la faisabilité d’une chimiothérapie intensive du sujet adulte < 60 ans (VANDA d’inspiration pédiatrique) ou âgé > 60 ans (EWALL) suivie du blinatumomab, avec un seul cas de CRS sévère, et ce malgré une toxicité hématologique accrue. En outre, tous les patients ayant pu bénéficier d’une allogreffe hématopoïétique au décours conservent une RMM persistante à long terme. La stratégie visant à obtenir une cytoréduction via une chimiothérapie de rattrapage intensive n’a pas permis de diminuer l’incidence de CRS ou de complications neurologiques par rapport aux données de la littérature.
Conclusion. L’utilisation d’une chimiothérapie intensive type VANDA à visée cytoréductive suivie du blinatumomab en monothérapie permet l’obtention d’une rémission complète chez 88 % des patients adultes atteints de LAL-B Ph- en R/R de notre centre avec un profil de tolérance acceptable chez une population âgée pour cette hémopathie. Les résultats obtenus chez nos patients ayant bénéficié d’une allogreffe de cellules souches en fait une alternative attrayante en situation de R/R en pont vers la transplantation.
06-96
Les leucémies aiguës myéloïdes à core binding factor : expérience du service d’hématologie et oncologie pédiatrique Casablanca (Maroc)
D. Dassouli*
Hématologie oncologie pédiatrique, Centre Hospitalier Universitaire Ibn Rochd, Casablanca, Maroc, Casablanca, Maroc
Introduction. Les leucémies aiguës myéloïdes à core binding factor (LAM-CBF) sont un groupe de leucémie reconnu comme associé à un pronostic favorable. Elles représentent 15 % des LAM de l’adulte regroupant les LAM codant pour deux anomalies récurrentes, t(8;21) et inv(16). L’addition d’autres anomalies cytogénétiques ne semble pas modifier le pronostic favorable de ce groupe de leucémie. La rémission complète des CBF atteint les 87 à 88 % avec une survie sans rechute estimée à 42 % à 10 ans. Même en cas de rechute, les LAM-CBF restent relativement de bon pronostic avec une survie à cinq ans d’environ 40 %.
Patients et méthodes. Il s’agit d’une étude rétrospective menée sur six ans et demi (janvier 2011 à juin 2017) au service d’hématologie et oncologie pédiatrique de Casablanca. Le diagnostic de la LAM a été retenu sur une étude de la cytologie, de l’immunophénotypage et du caryotype avec recours à la FISH en cas de caryotype normal. Ont été exclus de l’évaluation thérapeutique et pronostique les cas où le traitement n’a pas pu être démarré. Les patients ont été inclus au protocole AML 11. La rémission complète était définie par la présence de moins de 5 % de cellules blastiques au myélogramme après la deuxième induction.
Résultats. Sur la période d’étude, une série de 80 cas de LAM-CBF a été colligée dont sept patients ont été exclus de l’évaluation thérapeutique et pronostic. La LAM avec t(8;21) représentait 81 % des cas. La médiane d’âge était de 33 ans et des extrêmes allant de 3 ans à 57 ans. La prédominance était masculine avec un sex-ratio de 1,4. Le chlorome était noté chez quatre patients. L’anomalie cytogénétique a été détectée par caryotype conventionnel dans tous les cas sauf un où l’inversion du 16 a été détectée par FISH. L’anomalie cytogénétique était isolée dans 41 % des cas des t(8;21) et dans 60 % des cas des inv(16). La LAM était dans 60 % des cas de type 2 dans les t(8;21) et dans 87 % des cas de type 4 dans les inv(16). L’hyperleucocytose était retrouvée dans 21 % des cas. 15 patients, soit 20 % sont décédés au cours de la première induction. La rémission complète a été obtenue dans 41 % des cas et la rechute est survenue dans 19 % des cas après la fin de traitement. Sur quatre échecs (5 %) postinduction 1, un seul patient avec inv(16) a pu être rattrapé et mis en rémission complète.
Conclusion. L’évaluation de la réponse moléculaire dans les LAM-CBF permet d’améliorer encore plus leurs pronostics dans la mesure où l’allogreffe prend sa place en cas de réponse insuffisante. Bien que la t(8;21) et l’inv(16) soient généralement regroupées, il existe des différences dans leur présentation clinique ainsi que dans la réponse au traitement.
06-97
Leucémie aiguë promyélocytaire : une série de 63 cas au sein du service d’hématologie clinique et oncologie pédiatrique de Casablanca (Maroc)
H. Mouayad*1, D. Dassouli2
1 Service d’hématologie clinique et oncologie pédiatrique, Hôpital 20-Août, Casablanca, Maroc ; 2 Hématologie oncologie pédiatrique, Centre Hospitalier Universitaire Ibn Rochd, Casablanca, Maroc, Casablanca, Maroc
Introduction. La leucémie aiguë promyélocytaire (LAM3) est une entité rare de leucémie aiguë myéloïde caractérisée par la translocation (15 ; 17) (q22 ; q12) ; équivalente au réarrangement PML-RARA, au diagnostic une forte tendance hémorragique et un très bon pronostic à cause de sa sensibilité aux agents induisant une différenciation des blastes leucémiques tels que l’acide tout-trans-rétinoïque et le trioxyde d’arsenic. L’objectif de ce travail est de décrire le profil diagnostique et évolutif de cette pathologie dans notre centre d’hématologie.
Patients et méthodes. Une étude rétrospective avait été conduite sur la période allant de janvier 2003 à décembre 2017. Avaient été inclus tous les cas d’APL confirmés soit par la t(15;17)(q22;q12) sur le caryotype médullaire soit par la mise en évidence du gène de fusion PML-RARA en biologie moléculaire. La sévérité du syndrome hémorragique avait été évaluée selon le score hémorragique de l’OMS. Le score pronostic GIMEMA/PETHEMA avait été adopté. Les patients avaient été traités suivant le protocole ci-après. Induction : daunorubicine 60 mg/m2/j (J1-J3) ; Ara-C 200 mg/m2/j (J1-J7) ; ATRA 45 mg/m2/j (J1-J 45). Consolidation I : daunorubicine : 60 mg/m2/j (J1-J3) Ara-C 200 mg/m2/j (J1-J7), ATRA 45 mg/m2/j (J1-J15) Consolidation II : daunorubicine : 45 mg/m2/j (J1-J3) Ara-C 1 000 mg/m2/12 hrs (J1-J4), ATRA 45 mg/m2/j (J1-J15) Entretien (2 ans) : ATRA : 45 mg/m2/j (J1-J15) tous les trois mois + méthotrexate : 15 mg/m2/semaine + 6-mercaptopurine : 90 mg/m2/j. La prophylaxie du SNC était systématique. La dexaméthasone était administrée à raison de 10 mg/12 h pendant 5 jours pour prévenir le syndrome de différenciation. Le traitement de la CIVD était fait par transfusion de plasma frais congelé.
Résultats. 63 patients avaient été inclus avec un âge médian de 34,05 ans (1-60 ans) et un sex-ratio de 1,5. La médiane d’inclusion par an était de 4 patients. 47 patients (74 %) avaient un syndrome hémorragique, 24 (38 %) avaient une anémie et 11 (17 %) avaient un syndrome infectieux. 20 patients (31 %) avaient un syndrome hémorragique de grade 1, 8 (12 %) de grade 2, 11 (17 %) de grade 3 et 8 (12 %) de grade 4. Le nombre médian de leucocytes était de 12 G/L (0,2-92 G/L), le nombre médian de plaquettes était de 33 G/L (3-200 G/L), le taux médian d’hémoglobine était de 8,1 g/dL (4,3-14 g/dL). Le caryotype avait été réalisé au diagnostic chez 95 % des patients alors que la biologie moléculaire n’avait été réalisée que chez 36 % des patients. 34 % des patients avaient une CIVD. Au plan pronostique, 26 % des patients étaient classes haut risque, 59 % risque intermédiaire et 14 % faible risque. 10 patients (15 %) étaient décédés avant tout traitement par hémorragie. Après l’induction, 44 (70 %) patients étaient en rémission complète, 2 (3 %) en échec, 6 étaient décédés (9 %) dont 2 par choc septique, 2 par hémorragie cérébrale et 2 par hémorragie alvéolaire. 58 % des patients avaient reçu leur traitement d’entretien sans l’ATRA. Un cas de rechute avait été noté.
Conclusion. Les APL constituent un groupe rare de leucémie aiguë dans notre contexte. Les résultats de leur traitement sont encourageants mais reste à être amélioré notamment la prise en charge au diagnostic.
06-98
Les leucémies aiguës à cuplike nuclei : aspects morphologiques et moléculaires
F. Harieche*1, F. Belhadri2, H. Moussaoui3, F. Zerhouni1, RM. Hamladji4, R. Ahmed Nacer4
1 Service d’Hématologie-Greffe de Moelle Osseuse, centre Pierre et Marie Curie, Alger Centre, Algérie ; 2 Hématologie, centre Pierre et Marie curie Alger, Algiers, Algérie ; 3 Cpmc, CPMC, Sidi M’Hamed, Algérie ; 4 Service d’hématologie et greffe de moelle osseuse, Centre Pierre et Marie Curie d’Alger, Alger, Algérie
Introduction. Les leucémies à cuplike nuclei sont des leucémies aiguës myéloblastiques (LAM) dont les blastes présentent une invagination nucléaire en coupelle (cuplike).
Cet aspect semble caractériser les LAM avec des mutations du gène NPM1 (entité provisoire de la classification OMS) et est fréquemment associé à des mutations de FLT3.
L’objectif de notre étude est de vérifier localement la corrélation entre la morphologie de type cuplike (CN+) et la présence de la mutation du gène NPM1 et/ou FLT3.
Patients et méthodes. Notre étude inclut 21 patients (11 hommes et 10 femmes), atteints de LAM de Novo, avec mutation du gène NPM1 et/ou FLT3, diagnostiqués et suivis au service d’hématologie-greffe de moelle osseuse de l’EHS Pierre et Marie Curie.
Pour chaque patient, les blastes CN+ ont été identifiés puis quantifiés ; la mutation du gène NPM1 a été recherchée par PCR Quantitative en Temps réel (NPM1 Mutascreen, Ipsogen) ; la mutation FLT3-ITD a été recherchée par PCR qualitative (Seegene).
Les résultats obtenus ont été confrontés aux données cliniques et biologiques de ces patients.
Résultats. Les blastes CN+ ont été retrouvés chez 18/21 (85 %) des patients à des taux allant de 3 à 49 %. L’observation des LAM cuplike est plus fréquente dans les LAM peu différenciées ou à différenciation granuleuse LAM0, LAM1, LAM2 (14/21 soit 67 %) avec au diagnostic une fréquence plus élevée des hyperleucocytoses et une blastose importante.
La proportion des blastes CN+ dans la population de leucémies NPM1 muté est significativement plus importante que dans la population FLT3-ITD (86 % vs 42 % respectivement), ce qui suggère une association de la morphologie cuplike à la mutation NPM1.
Le profil immunologique des LAM CN+ présente des similitudes avec les leucémies aiguës promyélocytaires (LAP) : absence d’expression du CD34 et du HLA-DR.
Conclusion. Les LAM CN+ est une forme rare dont la fréquence est probablement sous-estimée car il s’agit d’une entité mal connue et peu décrite.
La cytologie CN+ est fortement prédictive d’anomalies moléculaires touchant NPM1 et FLT3.
06-99
Méthylation aberrante de l’ADN impactant l’expression des gènes HOX dans les cellules stromales mésenchymateuses de la moelle osseuse de syndromes myélodysplasiques et de leucémies aiguës myéloblastiques de novo
B. Roux1, F. Picou1, C. Vignon1, C. Debeissat2, N. Gallay2, M. Koubi1, P. Hirsch3, F. Mazurier1, F. Rouleux-Bonnin1, F. Gouilleux1, M. Maigre4, M. Hunault-Berger5, F. Delhommeau3, E. Gyan6, J. Domenech1, O. Herault*2
1 CNRS ERL 7001 lnox, Faculté de Médecine de Tours, Tours ; 2 Service d’hématologie biologique, CHRU de Tours, Tours ; 3 CDR Saint-Antoine, Université Pierre et Marie Curie, Paris ; 4 Hématologie, CH Chartres, Chartres ; 5 Service des maladies du sang, CHU - CHU Angers, Angers ; 6 Service d’hématologie et thérapie cellulaire, CHRU de Tours, Tours
Introduction. La méthylation de l’ADN contribue à la physiopathologie des hémopathies malignes et les agents hypométhylants sont couramment utilisés pour traiter les syndromes myélodysplasiques (SMD) et les leucémies aiguës myéloïdes (LAM). Dans ces maladies, l’état de méthylation de l’ADN des cellules stromales mésenchymateuses (CSM) de la moelle osseuse, susceptible de refléter leur fonctionnalité, pourrait être pertinent pour comprendre leur contribution à la physiopathologie.
Matériels et méthodes. Les CSM de patients atteints de SMD (n = 19 ; étude MYLESYM) et de LAM de novo (n = 21 ; FILOthèque LAM) ont été comparées à des CSM de moelles normales (n = 32). Elles ont été caractérisées par une quantification de la 5-méthylcytosine (dot-plots), des profils d’expression génique (qRT-PCR) de régulateurs clés de la méthylation (DNMT1, UHRF1, DNMT3A, DNMT3B, TET1, TET2, TET3, IDH1, IDH2), une identification des sondes méthylées de manière différentielle (DMP) et des régions (DMR) par méthylome array (850 000 sites), et une quantification de l’expression des gènes couplés au DMR (qRT-PCR).
Résultats. Les CSM de SMD et de LAM présentaient une hypométhylation globale de l’ADN et une sous-expression de DNMT1 et de UHRF1. L’analyse du méthylome a révélé un profil aberrant de méthylation dans les CSM de tous les SMD et d’un sous-groupe de LAM, qui a été objectivé surtout dans des gènes homéotiques, en particulier de la famille HOX dont HOXA1, HOXA4, HOXA5, HOXA9, HOXA10, HOXA11, HOXB5, HOXC4 et HOXC6. En outre, ces anomalies de méthylation étaient associées à un impact sur leur expression.
Conclusion. Ces résultats mettent en évidence des modifications de la méthylation de l’ADN dans les CSM de SMD et de LAM, tant au niveau global que focal, en dérégulant l’expression des gènes HOX bien connus pour leur implication dans la leucémogenèse. Une telle méthylation de l’ADN dans les CSM pourrait être la conséquence de la maladie maligne ou pourrait participer à son développement.
06-100
Inhibition du facteur de transcription leucémogène HOXA9 : validation dans un modèle leucémique murin
S. Depauw1, M. Lambert1, S. Jambon1, A. Paul2, DW. Boykin2, WD. Wilson2, MH. David-Cordonnier*3
1 Umrs1172, UMRS1172, Lille ; 2 Department of chemistry, Georgia State University, Atlanta, États-Unis ; 3 Inserm, UMR s 1172, Lille
Introduction. Le facteur de transcription HOXA9 est impliqué dans un grand nombre de leucémies aiguës myéloïdes (LAM) et d’autres hémopathies malignes (leucémies aiguës lymphoïdes, myélodysplasie, myélome multiple…). Ainsi, il est surexprimé dans toutes les leucémies de lignées mixtes à réarrangements du gène mll, les translocations MYST3-CREBP), NUP98-NSD1), ou de RUNX/AML1, ainsi que les mutations de NPM1 associées. Il a été montré qu’HOXA9 où il est nécessaire à la transformation leucémique par blocage de la différenciation normale et au maintien du caractère « progéniteur » des cellules leucémiques. Son activité leucémogène est directement reliée à sa capacité d’interaction à l’ADN. Aussi, nous avons sélectionné des composés inhibant cette interaction à l’ADN et montré leur activité in vitro et sur modèle leucémique murin de cellules de moelle osseuse de souris transformées par HOXA9.
Matériels et méthodes. Les approches moléculaires de sélection des composés ont été entreprises par approche dérivée de l’ELISA, retard en gel, empreinte à la DNase 1 et résonance plasmonique de surface. Une étude de modélisation moléculaire in silico a également été entreprise. Au niveau cellulaire, l’efficacité des composés a été validée par tests d’expression de la luciférase sous le contrôle des éléments de réponse à HOXA9. Les effets cellulaires ont été abordés par analyse transcriptomique et qRT-PCR puis validés par cytométrie en flux.
Résultats. Par ELISA et retard en gel, les composés ont été sélectionnés pour leur capacité à inhiber l’interaction à l’ADN. Cette interaction a été localisée plus précisément par empreinte à la DNAse 1 au niveau du site consensus d’HOXA9 et l’affinité des composés a été mesurée par technologie BIAcore (SPR). Les points de contacts précis des inhibiteurs sélectionnés ont été mis en évidence par analyse in silico de modélisation moléculaire. Ces composés sont capables d’inhiber la transcription de l’ADN médiée par HOXA9 comme montrée par test de luciférase.
L’évaluation des effets cellulaires a été effectuée sur une lignée cellulaire murine issue de cellules de moelle osseuse de souris transformées par le facteur de transcription HOXA9. Une analyse transcriptomique par traitement de ces cellules par un inhibiteur sélectionné a révélé l’inhibition de gènes associés avec les processus de mort cellulaire et de différentiation des cellules hématopoïétiques.
Enfin, nous avons montré par cytométrie en flux que ces composés sélectionnés induisent la mort de cette lignée leucémique HOXA9-dépendante, et par test clonogéniques qu’ils induisaient une différenciation des cellules de moins différenciées (CFU-GM) et colonies plus différenciées (CFU-G ou CFU-M).
Conclusion. Ainsi, les composés sélectionnés sont des inhibiteurs de la fonction oncogène d’HOXA9 par action sur sa capacité d’interaction à l’ADN. Ils représentent une stratégie prometteuse de ciblage d’HOXA9 dans les sous-types de leucémies HOXA9-dépendantes telles que les réarrangements MLL, les mutations de NPM et bon nombre d’autres altérations moléculaires.
06-101
Pachydermopériostose compliquée de leucémie aiguë myéloïde
S. Bouallegue*1, I. Fessi2, R. Sallemi3, L. Bouhajja3, M. Parrens4
1 hématologie clinique, hôpital régional de Zaghouan, Zaghouan, Tunisie ; 2 Médecine, hôpital régional Zaghouan, Zaghouan, Tunisie ; 3 Laboratoire d’anatomopathologie, hôpital régional de Zaghouan, Zaghouan, Tunisie ; 4 Département de pathologie, CHU de Bordeaux, Bordeaux
Introduction. La pachydermopériostose est la forme primitive, idiopathique de l’ostéoarthropathie hypertrophiante (OAH est une maladie héréditaire rare qui se transmet sur un mode autosomique dominant avec une pénétrance variable). Elle de manifeste principalement par des signes dermatologiques et des symptômes rhumatologiques. Des myélofibroses peuvent grever le pronostic. Nous rapportons une observation de pachydermopériostose compliquée de la survenue d’une leucémie aiguë myéloïde.
Résultats. Un homme de 60 ans présentait une pachydermopériostose (PDP) depuis l’âge de 30 ans. Cliniquement, le patient présente les signes cutanés et rhumatologiques de la PDP, avec hyperhydrose palmoplantaire, une hypertrophie des extrémités (mains, pieds, tiers inférieurs des jambes et des avant-bras), un hippocratisme digital au niveau des doigts et des orteils. L’examen cutanéomuqueux notait une pachydermie des extrémités avec un épaississement plissé, et une pilosité rare. Depuis septembre 2016, le patient consulte fréquemment aux urgences pour un syndrome anémique, associé à des diarrhées et des arthralgies. Au bilan biologique, L’hémogramme montre une pancytopénie avec des GB : 2700/mm, Hb : 5,3, VGM : 87 et des Plq : 68 000/mm. Il s’agit d’une anémie normochrome normocytaire arégénérative associée à une leucopénie et une thrombopénie. Un complément d’exploration par myélogramme met en évidence une moelle diluée à 2 reprises. La biopsie ostéomédullaire était en faveur d’une myélofibrose grade 3 selon Bauermeister. L’évolution a été marquée par l’aggravation de l’état général, des cytopénies et la survenue d’infections à répétition. L’exploration par imagerie a montré de nombreux foyers d’hématopoïèse extra-médullaire. Une BOM de contrôle a mis en évidence une transformation en leucémie aiguë myéloïde. L’évolution a été rapidement fatale malgré l’introduction d’une chimiothérapie à base de cytarabine à faible dose.
Conclusion. La pachydermopériostose est une maladie héréditaire dont le diagnostic repose sur l’association d’une arthropathie, d’un hippocratisme digital, d’une périostose, d’une pachydermie. C’est une entité rare. Les complications hématologiques ne sont exceptionnelles.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International