Annales de Biologie Clinique
MENUAnalyse microbiologique des solutions de contrôle d’endoscope : apport de la démarche d’accréditation dans la mise en application de la réglementation Volume 77, numéro 5, Septembre-Octobre 2019

L’endoscopie est un acte invasif associé à un risque infectieux le plus souvent d’origine endogène, lié au patient et à sa flore, sinon d’origine exogène, lié aux patients précédents ou à des équipements contaminés [1]. Les infections d’origine endogène sont associées à l’acte et ne peuvent être prévenues par la maîtrise des procédures de désinfection. Le risque d’infection exogène est considéré comme très faible car évalué à 1 cas pour 1,8 à 10 millions d’actes pour les endoscopies digestives [2, 3]. Entre 1966 et 1992 soit en 26 ans, seuls 281 cas de patients infectés après endoscopie digestive ont été rapportés aux États-Unis [2]. La iatrogénie infectieuse après endoscopie digestive a néanmoins été évaluée à 6 % dans une étude menée en 1988 et regroupant 116 hôpitaux américains [4], ce qui suggère que les données précédentes étaient largement sous-estimées. L’impact économique des infections d’origine exogène ne semble par ailleurs pas négligeable : une épidémie de six septicémies à Pseudomonas aeruginosa consécutives à une cholangio-pancréatographie rétrograde endoscopique (CPRE) aurait engendré un surcoût estimé à 206 273 $ [5]. Les cas cliniques d’infection exogène après endoscopie sont attribués le plus souvent à du matériel défectueux ou à des défaillances dans l’entretien des appareils : les principales causes d’une trentaine d’épidémies d’infections associées aux endoscopies digestives survenues aux Etats-Unis entre 1974 et 2004 concernaient des décontaminations inadéquates et des défauts de fabrication [6]. On estime que plus de 91 % des infections exogènes pourraient être évitées par une meilleure mise en œuvre des contrôles de qualité dans la fabrication et la décontamination des appareils [1].
Destinée à contrôler les pratiques de désinfection des endoscopes souples thermosensibles avec canaux, l’analyse des solutions de contrôle d’endoscopes a d’abord été définie par l’évaluation du niveau de contamination par le dénombrement de la flore mésophile totale aérobie revivifiable (FMTAR) et la détection de micro-organismes indicateurs d’un dysfonctionnement (MOID) de l’entretien de l’appareil [7]. Une liste de sept types de MOID était citée, et des niveaux cibles, d’alerte et d’action définies. D’autres recommandations et un texte règlementaire ont complété cette démarche en 2016 [8, 9]. Le dénombrement de la FMTAR comme la détection des MOID sont des étapes critiques car tout résultat erroné peut engendrer une sur- ou une sous-évaluation du risque infectieux résiduel après entretien de l’appareil, pouvant entraîner la privation non justifiée d’un acte endoscopique pour un patient ou l’exposition d’un ou plusieurs patients à une transmission croisée de micro-organismes à potentialité pathogène. Le contrôle microbiologique des solutions de contrôle d’endoscope doit donc respecter une démarche qualité permettant de garantir une fiabilité maximale dans la qualité de ses résultats.
Dans notre établissement, cette analyse était effectuée depuis 2006 par l’Unité fonctionnelle d’hygiène hospitalière et de lutte contre les infections nosocomiales (UFHHLIN). Elle consistait à dénombrer la FMTAR et un seul MOID, P. aeruginosa, et à interpréter les résultats uniquement selon des niveaux d’action à 100 unités formant colonie (UFC)/100 mL de solution de contrôle pour la FMTAR et 1 UFC/100 mL pour P. aeruginosa. Le mode opératoire et les niveaux cible, d’alerte et d’action n’avaient pas été révisés lors de la parution des référentiels en 2007 et 2016. Ce décalage nous a conduits à adopter une démarche qualité avant de présenter l’analyse à l’accréditation. Cette démarche volontaire avait pour but d’améliorer la qualité de la prise en charge des patients en atteignant les objectifs suivants : 1̊) répondre aux exigences réglementaires, 2̊) maîtriser le risque infectieux en endoscopie et 3̊) obtenir un niveau maximal de confiance dans la qualité des résultats.
Matériel et méthode
Présentation de l’hôpital
Notre établissement est un hôpital public d’environ 1 100 lits de médecine-chirurgie-obstétrique accueillant plus de 80 000 admissions par an. L’activité endoscopique avec des endoscopes souples avec canaux est exercée aux explorations fonctionnelles digestives, aux explorations fonctionnelles de pneumologie, en réanimation adulte et au bloc opératoire.
L’UFHHLIN : organisation et activité
L’UFHHLIN exerce les activités réglementaires de toutes les équipes opérationnelles d’hygiène et celles du laboratoire d’hygiène hospitalière. Celui-ci, créé en juin 2006, prend en charge trois vigilances : 1̊) Vigilance environnementale avec le contrôle microbiologique des eaux, des solutions de contrôle des endoscopes et des surfaces des services à risque infectieux de niveau 4 ; 2̊) Vigilance alimentaire avec le contrôle microbiologique du lait maternel ; 3̊) Vigilance épidémiologique avec la recherche de bactéries multirésistantes (BMR) ou hautement résistantes aux antibiotiques émergentes (BHRe) à partir de prélèvements à visée épidémiologique. Un système de management de la qualité (SMQ) respectant les critères d’exigence de la norme NF EN ISO/CEI 17025:2005 [10] a été déployé pour toutes les activités y compris pour les solutions de contrôle d’endoscope, prises en charge par le laboratoire depuis 2006. L’UFHHLIN est accréditée depuis 2012 pour le prélèvement et l’analyse de recherche et dénombrement des légionelles dans l’eau (section « Laboratoires », portée flexible de type A2), pour le contrôle microbiologique du lait maternel depuis 2015 (section « Santé humaine », portée flexible étendue de type B), pour le dépistage des entérocoques résistants aux glycopeptides (ERG, section « Santé humaine », portée flexible étendue de type B) depuis 2016, et pour le dépistage des entérobactéries productrices de carbapénèmase (EPC, section « Santé humaine », portée flexible étendue de type B) depuis 2017.
SMQ de l’UFHHLIN
La démarche qualité de l’UFHHLIN est pilotée en collaboration avec la responsable assurance qualité (RAQ) de l’établissement et implique l’ensemble de l’équipe. Des réunions qualité bimensuelles sont organisées depuis novembre 2010, avec la participation de certains membres de l’UFHHLIN : praticiens hygiénistes, cadre de santé, techniciens, secrétaire médicale, ainsi que la RAQ de l’établissement, un membre de l’atelier biomédical et un représentant des services techniques. L’ordre du jour systématique comprend les points suivants : 1̊) Bilan sur l’avancée du système et suivi des plans d’action ; 2̊) Fiches de dysfonctionnement, réclamation et travaux non conformes en cours ; et 3̊) Bilan d’un processus de la cartographie. Le planning annuel des réunions programme le processus à réviser à chaque séance. Le manuel qualité et le programme annuel d’audits internes sont examinés et validés lors des réunions qualité. Une revue de direction annuelle est organisée depuis 2011. Les audits internes externalisés sur les parties management et technique ont été mis en œuvre depuis 2013.
Exploitation des accréditations antérieures
Les dispositions internes relatives à la gestion de la portée flexible étendue de type B ont été utilisées pour encadrer les modifications imposées par la mise en conformité sur toute la chaîne du processus opérationnel et préparer le dossier d’accréditation : un formulaire de gestion de la portée flexible a été renseigné en suivant la méthode des 5M afin d’élaborer le dossier de validation de méthode. Ces documents ont été validés en réunion qualité. Les données des dossiers de validation de méthode élaborés pour la recherche de S. aureus dans le lait maternel, et des entérocoques et des entérobactéries pour le dépistage des ERG et des EPC ont complété celui pour la détection de ces trois types de MOID.
Elaboration de contrôles de qualité interne
La norme NF EN ISO/CEI 17025 prescrit dans le paragraphe « 5.9 Assurer la qualité des résultats d’essai et d’étalonnage » le point suivant : « Le laboratoire doit disposer de procédures de maîtrise de la qualité pour surveiller la validité des essais et des étalonnages entrepris. … Cette surveillance doit être planifiée et revue et peut inclure, sans s’y limiter, les éléments suivants : a) utilisation régulière de matériaux de référence certifiés et/ou d’une maîtrise de la qualité interne à l’aide de matériaux de référence secondaires ; … ».
Pour répondre à ce point, un contrôle de qualité interne (CQI) qualitatif de détection des MOID a d’abord été élaboré à partir de cinq souches : S. aureus ATCC 29213, Escherichia coli ATCC 25922, Enteroccocus faecalis (souche clinique), P. aeruginosa ATCC 27853et Candida albicans WDCM 00055. Une suspension de chaque souche à 0,5 unité Mc Farland a été diluée six fois en cascade au 1/25e dans du tampon diluant neutralisant pharmacopée (DNP, Oxoid®, Dardilly, France) avant filtration de chaque dilution en trois exemplaires sur membrane de nitrocellulose de porosité 0,45 μm (Millipore®, Molsheim, France). La membrane a été transférée sur gélose TSA (Millipore®), incubée à 30 ± 2 ̊C en aérobiose avec lecture à 48 heures, 72 heures et jusqu’à cinq jours en cas de culture stérile. La dilution retenue pour chaque souche était celle pour laquelle les UFC étaient dénombrables, entre 20 et 150 UFC/boite.
Après la première visite d’évaluation, un CQI pour validation des membranes filtrantes a également été élaboré en utilisant une souche d’E. faecalis WDCM 00009 (Sigma-Aldrich®, Buchs, Suisse) en tant que matériau de référence certifié (MRC). L’utilisation d’un CQI MRC pour le contrôle de la qualité des références et des lots de membrane filtrante était indispensable pour répondre aux critères d’exigence de la norme : son titre bactérien est garanti par le fabricant.
Évaluation des incertitudes de mesure
La norme NF EN ISO/CEI 17025 prescrit dans le paragraphe « 5.4.6. Estimation de l’incertitude de mesure » le point suivant : « Les laboratoires doivent aussi posséder et appliquer des procédures pour estimer l’incertitude de mesure. Dans certains cas, la nature de la méthode d’essai exclut un calcul rigoureux, métrologiquement et statistiquement valide, de l’incertitude de mesure. Dans de tels cas, le laboratoire doit au moins tenter d’identifier toutes les composantes de l’incertitude et faire une estimation raisonnable, tout en assurant que la manière d’en rendre compte ne donne pas une impression erronée de l’incertitude. Une estimation raisonnable doit se baser sur une connaissance de la performance de la méthode et sur le domaine de la mesure et faire appel, par exemple, à l’expérience acquise et aux données de validation antérieures ».
Pour répondre à ce point de la norme, deux actions ont été mises en œuvre : 1) une analyse de risque a été effectuée pour tenter d’identifier toutes les composantes de l’incertitude ; et 2) un biais a été calculé pour tenter de faire une estimation raisonnable. L’UFHHLIN participe depuis 2015 à l’évaluation inter laboratoire (EIL) organisée par l’Association générale des laboratoires d’environnement (AGLAE) pour les solutions de contrôle d’endoscopes. A chaque campagne, un z-score a été calculé par AGLAE à partir des résultats des laboratoires participants. À partir de ces résultats, le biais du laboratoire a été calculé par l’UFHHLIN en utilisant la formule suivante : biais en pourcentage = 100 x (moyenne obtenue par le laboratoire – moyenne interlaboratoire)/moyenne interlaboratoire.
Relations avec les clients
Les clients confiant à l’UFHHLIN l’analyse des solutions de contrôle d’endoscope étaient les quatre secteurs utilisant des endoscopes souples thermosensibles à canaux. Un groupe de travail a été constitué avec chacun pour la mise en conformité à l’instruction du 04 juillet 2016, et a effectué une analyse de risque pour déterminer les points critiques et les MOID liés à l’utilisation de chaque famille d’endoscopes. L’instruction du 04 juillet 2016 précise : « En pratique, il est recommandé … de vérifier l’absence de micro-organismes indicateurs d’un dysfonctionnement en identifiant les colonies apparues … les agents concernés sont les entérobactéries, Entérocoques, Pseudomonas aeruginosa et autres Pseudomonas, Stenotrophomonas maltophilia, Acinetobacter sp, Staphylococcus aureus et Candida sp, champignons filamenteux. ». L’utilisation du terme « il est recommandé » suggère que le respect strict de la liste des MOID mentionnés n’est pas obligatoire. En raison de la charge de travail, qui reviendrait à identifier chaque type de colonies isolées sur gélose trypticase soja, nous avons décidé d’exploiter les analyses de risque effectuées pour chaque famille d’endoscopes pour focaliser dans un premier temps sur les MOID les plus critiques. Ceux-ci ont été déterminés en concertation avec les cliniciens de chaque secteur d’endoscopie lors de la rédaction des analyses de risque. Une convention liant chaque secteur et l’UFHHLIN a été rédigée sur la base de ces résultats, notamment en listant les MOID à détecter spécifiquement pour chaque famille d’endoscopes.
Référentiels
L’instruction du 04 juillet 2016 relative au traitement des endoscopes souples thermosensibles à canaux au sein des lieux de soins était le seul texte réglementaire étudié [9], complété par les documents « Éléments d’assurance qualité en hygiène relatifs au contrôle microbiologique des endoscopes et à la traçabilité en endoscopie » du Comité technique des infections nosocomiales et des infections liées aux soins (CTINILS) et « Surveillance microbiologique de l’environnement dans les établissements de santé » du Centre de coordination des comités de lutte contre les infections nosocomiales du Sud-Ouest (C.CLIN Sud-Ouest) [7, 8]. Les autres référentiels étaient la norme NF EN ISO/CEI 17025 pour le management de la qualité et les documents Cofrac LAB REF 02, LAB INF 32, LAB REF 08, GEN REF 11 et LAB GTA 23 [10-15]. La norme ISO 7704 [16] a été utilisée pour le protocole de validation des membranes filtrantes.
Résultats
Étude des référentiels
Les résultats de l’étude des quatre référentiels abordant les aspects techniques de l’analyse sont détaillés dans le tableau 1. La plupart des critères d’exigence étaient similaires, quelques précisions étant apportées par certains. 1̊) Pour la liste des MOID, le CTINILS ne cite pas les entérocoques et les champignons filamenteux alors que le C.CLIN Sud-Ouest ne cite pas les entérocoques et réduit les champignons filamenteux aux Aspergillus, tout en mentionnant les causes possibles de détection des MOID : mauvais stockage, mauvais séchage, ou risque pour le patient. 2̊) Si une analyse immédiate n’est pas possible, l’échantillon doit être maintenu pendant une durée maximale de 24 heures à 4 ̊C pour le CTINILS, et à 5 ± 3 ̊C pour l’instruction. Pour le C.CLIN Sud-Ouest, elle peut être effectuée dans les deux heures, sinon l’échantillon doit être maintenu à 5 ± 3 ̊C pendant une durée maximale de 24 heures. Le CTINILS recommande que le transfert du prélèvement soit effectué dans un récipient réfrigéré maintenant l’échantillon à + 4 ̊C si l’analyse est effectuée dans une structure extérieure à l’établissement. 3̊) Le CTINILS et le C.CLIN Sud-Ouest mentionnent le degré de porosité des membranes de filtration, et le C.CLIN Sud-Ouest précise que les étuves doivent être à 30 ± 2 ̊C et recommande l’utilisation d’un poste de sécurité microbiologique pour les endoscopes de haut niveau. 4̊) Le rinçage de la membrane filtrante à l’eau stérile est recommandé par le CTINILS et le C.CLIN Sud-Ouest, qui mentionnent des volumes de rinçage différents. 5̊) La détection des MOID doit être confirmée par une galerie d’identification après réisolement pour le LAB INF 32. 6̊) Ce dernier précise que la portée d’accréditation est en portée fixe et selon une méthode interne. 7̊) Des niveaux cibles, d’alerte et d’action après désinfection de niveau intermédiaire avec rinçage à l’eau pour soins standard sont définis par le CTINILS et le C.CLIN Sud-Ouest.
L’étude du LAB GTA 23 avait été écartée dans un premier temps car les solutions de contrôle d’endoscope n’apparaissaient pas dans la liste des analyses microbiologiques des eaux citée dans son paragraphe 7.1. La révision 02 du document, applicable à compter du 15 juillet 2017, a intégré les solutions de contrôle des endoscopes dans cette liste. L’étude d’impact menée par l’UFHHLIN sur ce document concluait que pour le laboratoire seules les revues des demandes, appels d’offres et services aux clients étaient potentiellement modifiables, en termes de clarification avec le client sur le contexte réglementaire ou d’agrément dans lequel s’inscrivent les analyses. Aucune disposition relative aux exigences techniques n’était impactée.
Gestion de la portée flexible
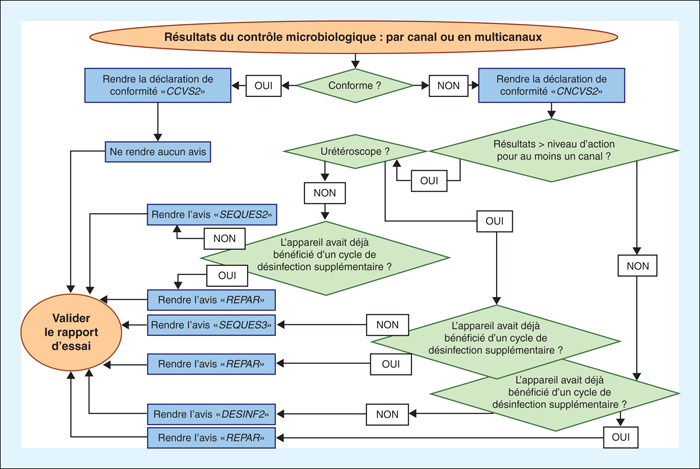
Un formulaire de gestion de la portée flexible a été renseigné afin de satisfaire aux critères d’exigence des référentiels. Le système d’information du laboratoire a été paramétré selon les critères du LAB INF 32 : technique d’analyse, volume injecté et recueilli, et expression du dénombrement de la FMTAR ou de la détection des MOID. Des déclarations de conformité existant déjà, quatre avis ont été définis pour tenir compte des nouveaux niveaux cibles, d’alerte et d’action, et pour que les praticiens hygiénistes puissent inclure dans les rapports d’essai la conduite à tenir en cas de résultats non conformes (tableau 2). Pour satisfaire aux exigences du LAB REF 02, cette définition a entraîné la modification des dispositions internes relatives à l’habilitation des signataires de rapport d’essai et un logigramme décisionnel a été créé pour encadrer l’émission des avis (figure 1). Les groupes de travail avec chaque secteur d’endoscopie réunis de juin à septembre 2017 ont élaboré 14 dispositions internes. Parmi celles-ci, les conventions prévoyaient qu’une réunion qualité ait lieu au minimum une fois par an pour effectuer les revues de contrat, de capacité, de rapport d’essai et des analyses de risques, et exposer l’exploitation des résultats des EIL. Les autres dispositions internes concernaient les CQI et les EIL, qui ont été introduits dans le maintien d’habilitation technique, et les critères d’exigence relatifs aux points critiques à observer, aux critères de conformité, et au maintien d’habilitation ont été précisés. Enfin, les dispositions internes concernaient également l’habilitation des signataires de rapports d’essai, sur le contrôle de connaissances pour les avis et les critères à observer, dont ceux relatifs à la politique d’utilisation de la marque Cofrac. Ce dernier avait été introduit dans les habilitations, afin d’éviter la lecture particulièrement fastidieuse du document Cofrac GEN REF 11. Aucun CQI quantitatif correspondant à la FMTAR n’a été trouvé dans le commerce pour la technique de filtration sur membrane et n’a donc pu être introduit dans les dispositions internes. Seuls des CQI qualitatifs ont été élaborés pour la détection des MOID, à partir de dilutions de suspensions bactériennes à 1/15 625e (trois dilutions en cascade au 1/25e) pour C. albicans, à 1/390 625e (quatre dilutions en cascade au 1/25e) pour S. aureus, E. coli, et E. faecalis, et à 1/781 250e (quatre dilutions en cascade au 1/25e puis une dilution au ½) pour P. aeruginosa. Ces CQI ont été utilisés en routine à partir de juillet 2017. Les modalités d’information des utilisateurs ont été décidées et tracées lors des réunions qualité.
Validation de méthode
Deux sous-processus ont été distingués : 1̊) la culture sur gélose et le dénombrement de la FMTAR et 2̊) l’identification bactérienne (tableau 3). La partie relative au premier a été renseignée à partir de l’analyse de risque, de la partie quantitative de l’exploitation des EIL et du calcul du biais. Celle relative au deuxième l’a été à l’aide de l’analyse de risque, de la partie qualitative de l’exploitation des EIL, et de la variabilité inter-opérateurs mise en œuvre lors des validations de méthode antérieures pour l’identification de S. aureus, des entérocoques et des entérobactéries. Les risques les plus critiques étaient l’erreur d’identité de l’endoscope, l’erreur de nature et de volume de l’échantillon adressé au laboratoire, et le défaut de maîtrise des compétences du personnel. Le contrôle de la stérilité des consommables, tampon DNP et flacon de prélèvement compris, a été organisé une fois par mois avec alternance de chaque secteur d’endoscopie à l’aide d’un blanc de prélèvement servant pour un essai à blanc. Le neutralisant dans le flacon de prélèvement pour neutraliser l’activité résiduelle des désinfectants présents dans le prélèvement a été considéré comme superflu. Ces désinfectants seraient le chlore si l’eau de rinçage des endoscopes était chlorée, et l’acide peracétique provenant de la désinfection précédente si les rinçages étaient insuffisants. Leurs quantités ont été considérées comme négligeables car : 1̊) en théorie il ne reste plus d’eau dans l’endoscope, puisqu’il a été désinfecté et rincé au minimum six heures avant le prélèvement et qu’il a été séché aussitôt après ; 2̊) si l’endoscope n’est pas totalement sec au moment du prélèvement, les quantités résiduelles d’eau et de désinfectant sont très faibles ; 3̊) conformément à la réglementation le tampon DNP contient 0,5 % de thiosulfate de sodium soit 0,5 mg pour 100 mL de tampon, ou par canal ou endoscope prélevé ; 4̊) dans l’hypothèse où le volume d’eau résiduelle serait de 10 mL, volume très élevé donc surestimé, le prélèvement filtré contiendrait 0,5 mg de thiosulfate pour 10 mL d’eau, soit 50 mg/L d’eau ; 5̊) il a été démontré pour le paramètre légionelles qu’une quantité de thiosulfate de sodium de 20 mg dans le flacon de prélèvement était suffisante pour neutraliser toute trace de chlore dans un litre d’eau en cas de surchloration. La concentration de 50 mg/L est donc suffisante pour gérer un risque d’eau résiduelle dans un endoscope mal séché, si elle se retrouvait intégralement dans le prélèvement de l’endoscope, ce qui est hypothétique. Il apparaît donc inutile de contrôler la quantité de thiosulfate de sodium dans le tampon DNP comme d’utiliser des flacons de prélèvement contenant une quantité supplémentaire de neutralisant. La participation à un programme d’EIL a été considérée comme satisfaisant à un contrôle de rendement. Le z-score se situait toujours entre -2 et +2. En l’absence de CQI quantitatif pour la FMTAR, les incertitudes de mesure ne pouvaient pas être calculées. Une analyse de risque sur l’essai a été élaborée selon la méthode des 5M afin d’identifier toutes les composantes de l’incertitude, et le calcul d’un biais mis en œuvre pour faire une estimation raisonnable. Les résultats se situaient entre -7,75 % et +16,25 %. Un contrôle des membranes filtrantes a été organisé en comparant les rendements obtenus après filtration à chaque changement de lot. Les CQI de détection des MOID ont été organisés selon une fréquence mensuelle, avec un type de MOID par mois. Les dispositions exigées par le document LAB GTA 23 pour le contrôle des conditions ambiantes comme l’accès au laboratoire étaient déjà mises en œuvre pour la recherche et le dénombrement des légionelles dans l’eau.
Visites d’évaluation
La demande d’extension d’accréditation a été adressée le 15 avril 2017 à la section « Laboratoires », qui a répondu que la portée flexible étendue de type B était impossible à utiliser en l’absence de référentiel interne à la section pour l’évaluation d’une activité de contrôle microbiologique des eaux selon ce type de portée. L’UFHHLIN a reformulé sa demande en portée FIXE, mais le formulaire de gestion de la portée flexible a été utilisé dans l’attente de la visite d’évaluation. L’UFHHLIN a présenté le dénombrement de la FMTAR et l’identification de trois types de MOID : S. aureus, entérocoques et entérobactéries. Les nouvelles dispositions internes sur le « Contrôle microbiologique des solutions de contrôle des endoscopes » et l’« Expression des résultats, déclarations de conformité et avis » ont été approuvées les 24 et 25 août 2017. La méthode en vigueur et la nouvelle à mettre en œuvre ont été comparées du 21 août au 1er septembre 2017. Les résultats démontrant que les nouvelles dispositions internes étaient maîtrisées de la réception du prélèvement à la diffusion des rapports d’essai, leur mise en application ont été prononcés le 4 septembre 2017. L’évaluation de surveillance pour la recherche et le dénombrement des légionelles dans l’eau et d’extension pour les solutions de contrôle d’endoscope a eu lieu les 14 et 15 septembre 2017. Une fiche d’écart non critique a été ouverte et deux points à surveiller ont été relevés (tableau 4). Le constat d’écart portait sur le contrôle des consommables et réactifs avant leur utilisation, dont celui des membranes de filtration qui ne respectait pas intégralement la norme ISO 7704 [16]. Chaque lot de membranes était contrôlé en le comparant au lot précédent, mais aucune comparaison de méthode entre les techniques d’étalement et de filtration, pour deux échantillons contenant une concentration équivalente en MRC, n’avait été effectuée. Les actions proposées pour clôturer la fiche d’écart étaient : 1̊) la révision de l’analyse de risque ; 2̊) l’installation et la mise en production d’un logiciel de gestion des stocks permettant de programmer les contrôles à réception sans attendre les CQI ; 3̊) la relecture de la norme ISO 7704 pour décider de son adoption ou non stricto sensu ; et 4̊) en fonction de cette décision, la révision de l’analyse de risque ou de la procédure de contrôle des membranes et – le cas échéant – la programmation de manipulations comparant les techniques d’étalement et de filtration et comprenant un MRC. Les deux points à surveiller concernaient la partie technique avec 1̊) un renforcement de la procédure d’habilitation initiale par l’utilisation de souches atypiques pour l’identification des MOID ; et 2̊) l’absence de certains critères de décision suite à contrôle au sujet de certains tests de validation. Les six points forts relevés par l’évaluateur sont mentionnés dans le tableau 4. L’accréditation a été prononcée par le Cofrac le 15 novembre 2017. Le plan d’actions a conduit à l’adoption stricto sensu de la norme ISO 7704 pour la validation des membranes de filtration et à la révision de la procédure pour cette validation. Les manipulations comprenant une souche d’E. faecalis WDCM 00009 certifiée ont été mises en œuvre du 24 au 30 janvier 2018, et ont conduit à la première validation des membranes de filtration conformément à tous les critères d’exigence de la norme ISO 7704.
La première visite d’évaluation de surveillance a eu lieu les 17 et 18 janvier 2019. Elle coïncidait avec une visite d’extension, le laboratoire ayant demandé de changer de type de portée, en passant de la portée FIXE à la portée FLEX3. Pour la première fois, cette visite portait sur l’intégralité des essais rendus sous accréditation par le laboratoire selon la norme NF EN ISO/CEI 17025, que ceux-ci dépendent de la section « Laboratoires » (légionelles et solutions de contrôle d’endoscope) ou de la section « Santé humaine » (lait maternel et dépistage des ERG et des EPC). La détection de Candida sp a été présentée à cette occasion. Trois fiches d’écart ont été ouvertes : deux relatives à la métrologie et une concernant un essai de recherche et dénombrement des légionelles dans l’eau. Aucune ne portait sur les solutions de contrôle d’endoscopes. Suite à cette visite, la portée FLEX3 a été accordée au laboratoire pour l’analyse de contrôle de désinfection des endoscopes. La détection de P. aeruginosa a été validée peu de temps après.
Les actions menées par rapport à l’accréditation de l’analyse microbiologique des solutions de contrôle des endoscopes sont résumées dans le tableau 5.
Discussion
À sa création en 2006, le laboratoire de l’UFHHLIN a repris les essais d’hygiène hospitalière pratiqués par le laboratoire de microbiologie clinique. Ce transfert a nécessité une priorisation des essais à réactualiser ou à créer en raison de priorités institutionnelles : internalisation de la recherche et du dénombrement des légionelles dans l’eau pour encadrer le déménagement sur un nouvel hôpital, reprise du contrôle microbiologique du lait maternel pour l’autorisation de fonctionnement du lactarium à usage intérieur, création des dépistages d’ERG et d’EPC pour les patients à risque de portage et notamment ceux de retour de l’étranger. Pour le premier dossier prioritaire, l’UFHHLIN s’est engagée dans une démarche qualité par la rédaction d’un manuel de management de la qualité recouvrant toutes ses activités [17]. Cette démarche globale, initiée par le contexte réglementaire du paramètre légionelles, a été exploitée pour les deux dossiers prioritaires suivants. L’accréditation du contrôle microbiologique du lait maternel ayant été attribuée à l’UFHHLIN selon la ligne de portée flexible étendue de type B [18], les dispositions internes de gestion de la portée flexible ont été améliorées. Elles ont été utilisées pour l’accréditation des dépistages d’ERG et d’EPC, notamment pour les extensions d’accréditation relatives aux antibiogrammes [19] et à l’amplification génique. L’obtention de la portée de type B pour ces dépistages confirmait la reconnaissance par le Cofrac des compétences de l’UFHHLIN en matière de développement de méthode.
Jusqu’en août 2017, l’analyse de contrôle microbiologique des solutions de contrôle d’endoscopes par notre laboratoire comprenait le dénombrement de la flore totale aérobie et de P. aeruginosa avec des niveaux d’action à 100 et 1 UFC/100 mL. Nous enregistrions 10 ans de retard dans la mise en application des recommandations et un an dans celle de la réglementation, moins d’un an avant la visite de certification HAS programmée en janvier 2018. Le risque infectieux lié à l’acte endoscopique était sous-estimé. Face à cette situation, le dénombrement des micro-organismes revivifiables à 30 ̊C par filtration sur membrane et la détection des S. aureus, des entérocoques et des entérobactéries ont été présentés dans un premier temps. La visite d’évaluation a conduit à un seul écart sur le contrôle des consommables et réactifs. L’écart était non critique car la membrane contrôlée même sans respecter intégralement la norme ISO 7704, et parce que tous les résultats des EIL étaient conformes, attestant de la capacité à rendre des résultats suffisamment proches de la moyenne de ceux des laboratoires participants sans dérive au cours du temps. Le contrôle des consommables et réactifs est imposé par le paragraphe « 4.6. Achat de services et de fournitures » de la norme NF EN ISO/CEI 17025. Dans son paragraphe « 7.9. Qualité des résultats d’essai », la révision 02 du document LAB GTA 23 cite les normes ISO 7704 pour le contrôle des membranes filtrantes et NF EN ISO 11133 [20] pour le contrôle des milieux de culture et réactifs, bien que leurs domaines d’application ne comprennent pas les solutions de contrôle d’endoscopes. Applicable à partir du 15 juillet 2017, sa mise en œuvre n’était pas obligatoire lors de la visite d’évaluation de septembre 2017. C’est parce que l’analyse de risque précisait que la norme ISO 7704 devait être respectée stricto sensu pour le contrôle des membranes qu’une partie du constat d’écart a été formulée. Par ailleurs, un contrôle de rendement devrait être mis en œuvre pour satisfaire aux exigences de la norme NF EN ISO 11133. En l’absence de CQI quantitatif représentatif de la FMTAR dans le commerce pour la technique de filtration sur membrane, nous avons choisi de mettre d’abord au point le contrôle de validation des références puis des lots de membranes filtrantes à partir d’une souche de E. faecalis MRC. Cette élaboration a été approuvée par les auditeurs lors de la visite de surveillance de 2019, car la labélisation du CQI en MRC permet de garantir la qualité du contrôle, et donc que chaque lot de membrane filtrante n’altère pas le dénombrement de la FMTAR. À terme, elle ouvre également la voie à l’élaboration d’un CQI quantitatif pour contrôle de rendement et d’une carte de contrôle permettant de calculer les incertitudes de mesure. Sur ce point, la norme NF EN ISO/CEI 17025 précise que « dans certains cas, la nature de la méthode d’essai exclut un calcul rigoureux, métrologiquement et statistiquement valide, de l’incertitude de mesure. Dans de tels cas, le laboratoire doit au moins tenter d’identifier toutes les composantes de l’incertitude et faire une estimation raisonnable, tout en assurant que la manière d’en rendre compte ne donne pas une impression erronée de l’incertitude ». Toutes les composantes de l’incertitude ont été identifiées par l’analyse de risque, et une estimation raisonnable a été effectuée en calculant un biais à partir des résultats des EIL. Ce biais s’appuyant sur l’écart observé par rapport aux données de validation de chaque campagne d’EIL, il a été admis par les auditeurs Cofrac qu’il répondait à ce point précis de la norme, dans l’attente de l’exploitation des résultats du CQI quantitatif. Enfin, l’instruction du 04 juillet 2016 précisant que « la solution de prélèvement doit […] neutraliser l’activité résiduelle des désinfectants (sinon risque de faux négatif) », un contrôle de la quantité de neutralisant spécifique du désinfectant présent dans le tampon DNP prêt à l’emploi aurait pu paraître nécessaire. L’analyse de risque a démontré qu’il était superflu et qu’il était inutile d’utiliser des flacons de prélèvement contenant du neutralisant.
L’expression des lignes de portée d’accréditation pour les analyses microbiologiques des eaux est définie par les documents LAB INF 32 et LAB REF 08. Le profil de flexibilité pour l’analyse des solutions de contrôle d’endoscope a été défini en portée FIXE dans la révision 01 du LAB INF 32, applicable à partir du 15 septembre 2017, ce document précisant que« le laboratoire est reconnu compétent pour pratiquer les essais en respectant strictement les méthodes mentionnées dans la portée d’accréditation. Les modifications techniques de la méthode ne sont pas autorisées. ». Cette mention correspond à celle utilisée par le LAB REF 08 pour les méthodes non reconnues. L’obligation d’utiliser la portée FIXE semble donc motivée par l’absence de méthode reconnue. Le LAB INF 32 précise que la référence de la méthode correspond à une « méthode interne (référence et version du mode opératoire à préciser) », sans citer aucun des référentiels étudiés dans ce travail. Pourtant, la fiche 8 de l’instruction du 04 juillet 2016 décrit précisément les critères d’exigence de l’analyse et pourrait correspondre au quatrième alinéa de la définition des méthodes reconnues du LAB REF 08 : « Une méthode rendue d’application obligatoire, si elle est décrite, ou référencée dans un texte réglementaire, dès lors que cette méthode est utilisée dans le contexte d’application obligatoire ». Par ailleurs, l’obligation d’accréditer les solutions de contrôle d’endoscopes selon la portée FIXE est potentiellement préjudiciable à tout laboratoire accrédité en cas de modification de la réglementation sur la technique d’analyse, celui-ci devant modifier sa méthode interne avant la visite d’évaluation suivante et rendre les résultats hors accréditation. Cette obligation n’apparaît pas dans le LAB REF 08, qui définit dans sa révision 04 les autres types de portée de la section « Laboratoires » : flexibles FLEX1, FLEX2 et FLEX3. La portée FLEX3 est compatible avec l’absence de méthode reconnue pour le contrôle microbiologique des endoscopes, puisque dans son cas « le laboratoire est reconnu compétent, dans le domaine couvert par la portée générale, pour adopter toute méthode reconnue et pour développer ou mettre en œuvre toute autre méthode dont il aura assuré la validation. », les portées FLEX1 et FLEX2 ne concernant que les méthodes reconnues. Les documents LAB INF 32 et LAB REF 08 sont donc contradictoires : ou bien l’instruction du 04 juillet 2016 est une méthode reconnue et toutes les portées sont applicables, ou bien elle n’est pas une méthode reconnue et les portées FIXE et FLEX3 sont applicables. Enfin, la révision 02 du LAB GTA 23, qui inclut les solutions de contrôle des endoscopes dans le domaine de la microbiologie appliquée aux analyses des eaux, n’aborde que les portées FIXE et FLEX1. Ce document ajoute donc à la confusion. Au-delà de ces contradictions, la portée FLEX3 étant similaire à la portée flexible étendue de type B définie par le SH REF 08 [21], et les compétences de l’UFHHLIN en gestion de la portée flexible étendue de type B et en développement de méthode étant reconnues par la section « Santé humaine » du Cofrac, l’impossibilité d’utiliser la portée FLEX3 apparaissait paradoxale. Sur la base de cet argumentaire, une demande d’extension pour modification de portée a été formulée au Cofrac, qui l’a accepté et la visite d’extension de janvier 2019 a conduit à l’obtention de la portée FLEX3 en lieu et place de la portée FIXE, justifiant a posteriori la demande initiale. Le maintien de la portée FIXE aurait potentiellement mis en difficulté le laboratoire, en cas de changement de réglementation entre deux visites d’accréditation. Dans ce cas le laboratoire aurait été contraint ou bien d’appliquer le texte réglementaire et de rendre les résultats hors accréditation, ou bien de ne pas appliquer le texte réglementaire pour pouvoir continuer à rendre les résultats sous accréditation. La portée flexible FLEX3 permet d’échapper à cette contrainte.
Afin de satisfaire en quelques mois aux critères d’exigence de la réglementation et des référentiels, améliorer la qualité de la prestation et s’assurer de la confiance des résultats de l’analyse des solutions de contrôle d’endoscopes, les résultats de la démarche initiée en 2010 pour l’accréditation du paramètre légionelles ont été exploités. Aucune disposition relative aux exigences techniques n’était impactée et les trois types de MOID présentés ne nécessitaient aucune manipulation supplémentaire à celles des validations de méthode antérieures ou de la mise en œuvre des EIL et des CQI. La détection d’un type de MOID rendu hors accréditation (Candida sp) a été présentée à la visite d’évaluation suivante, au moment de l’obtention de la portée FLEX3. La détection de P. aeruginosa a été validée après cette obtention, sans attendre la visite d’évaluation programmée en 2020. Il est prévu de valider celle des autres types de MOID au fur et à mesure des validations de méthode à partir des résultats des CQI et des EIL. La visite de certification HAS n’a conduit à aucune remarque, alors que les secteurs d’endoscopie ont tous été visités. Notre stratégie a donc eu les impacts recherchés, aussi bien pour la sécurité des patients que sur le plan institutionnel.
Liens d’intérêts
les auteurs déclarent ne pas avoir de lien d’intérêts en rapport avec cet article.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International