Hématologie
MENULa leucémie myélomonocytaire juvénile Volume 23, numéro 2, Mars-Avril 2017


hôpital Robert Debré,
service d’hématologie pédiatrique,
Paris, France
hôpital Robert Debré,
département de génétique,
Paris, France
- Mots-clés : leucémie myélomonocytaire juvénile, RAS
- DOI : 10.1684/hma.2017.1256
- Page(s) : 122-34
- Année de parution : 2017
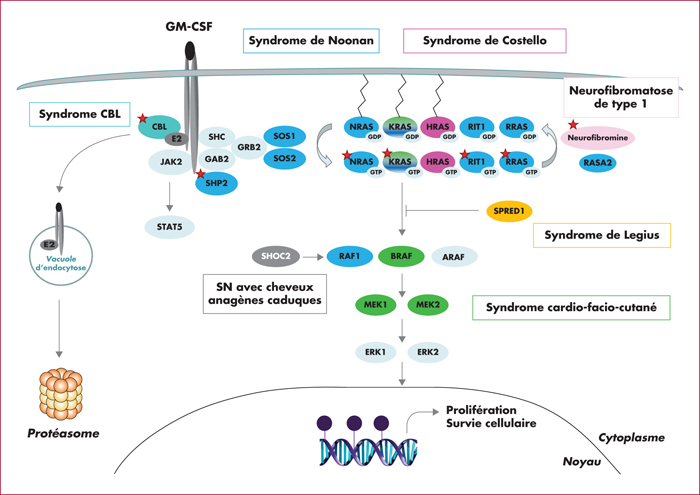
La leucémie myélomonocytaire juvénile (LMMJ) est un syndrome myélodysplasique/myéloprolifératif agressif de la petite enfance lié à une hyperactivation de la voie de signalisation RAS/MAP-kinase. Il s’agit d’une hémopathie rare et difficile à diagnostiquer du fait de la grande hétérogénéité des présentations cliniques et de l’absence de test diagnostique spécifique. Un tiers des LMMJ surviennent dans un contexte de syndromes prédisposants liés à une hyperactivation constitutive de la voie RAS, les RASopathies, qui nécessitent une prise en charge particulière. Il est donc indispensable, chez tout patient avec LMMJ, de mener une enquête génétique constitutionnelle, clinique et biologique. La meilleure connaissance des bases moléculaires des LMMJ a permis, ces dernières années, d’améliorer la stratification thérapeutique des patients, et il est donc aujourd’hui licite de proposer à certains patients, sur les bases de leur analyse moléculaire tumorale et constitutionnelle, une surveillance clinique et biologique simple. Cependant, un grand nombre de patients doivent toujours avoir recours à un traitement curateur. Le traitement de référence reste aujourd’hui la greffe de cellules souches hématopoïétiques qui permet de guérir environ 50 % des enfants. Les espoirs se concentrent sur les agents déméthylants et sur l’utilisation de thérapeutiques ciblées sur les voies de signalisation dérégulées dans la LMMJ.