Epileptic Disorders
MENUThe semiology of benign focal epilepsy with affective symptoms Volume 19, numéro 2, June 2017

Benign focal epilepsy with affective symptoms (BFEAS), first described by Dalla Bernardina et al. in 1980 (Dalla Bernardina et al., 1980), is a rare childhood epilepsy syndrome essentially characterized by “epileptic attacks with affective symptoms of a terrifying type”. Since the original description, approximately 50 cases of BFEAS have been reported. Presently, BFEAS is considered a clinical phenotype of benign childhood seizure susceptibility syndrome (BCSSS), sharing features with both Rolandic epilepsy and Panayiotopoulos syndrome. To our knowledge, however, none of the studies included video-EEG data. Herein, we detail the electroclinical features of a patient diagnosed with BFEAS, as well as provide video-EEG of one of his typical seizures with affective symptomatology.
Case study
A 9-year-old boy from Brazil was born to healthy, non-consanguineous parents after an uneventful pregnancy. Birth and psychomotor development were normal, and he never had febrile seizures. Family history was remarkable for one seizure (afebrile, of unknown characteristics) experienced by the patient's maternal aunt when one year old.
At the age of 2 years, the patient started having monthly episodes when he would awake from sleep (between three and five o’clock in the morning) to sit in bed and stare for a few minutes (two to four minutes) with his consciousness impaired. These paroxysms were accompanied with coughing, repeated swallowing, tachycardia, excessive sweating, and, occasionally, vomiting and urinary incontinence. Postictally, there was prominent fatigue.
At the age of 4 and a half years, he had his first convulsive seizure, which was, in fact, a status epilepticus. He awoke from sleep and had repeated swallowing, as well as opposition of the right thumb against the second finger, followed by secondary generalization. The seizure lasted approximately 25 minutes and was not aborted until he received anticonvulsant medication at the local emergency department. He had significant postictal fatigue and headache. The patient underwent brain MRI, which was found to be unremarkable; he was then discharged on oxcarbazepine. Thereafter, he continued to have multiple episodes of status epilepticus (of the same semiology described above) at a frequency of one every 20 days, despite therapy with lamotrigine, valproate, and diazepam. The secondary generalized seizures persisted until the age of 5 and a half years, when sulthiamine was added to this antiepileptic drug regimen.
At the age of 6 years, he started having monthly events characterized by sudden awakening from sleep, intense fear, and excessive sweating in association with impaired consciousness. He would scream asking for his mother, whom he would hug as soon as she arrived to console him. These episodes lasted for five to seven minutes, and were followed by fatigue. Interestingly, all his seizures, throughout his life, have occurred during sleep and between three and five o’clock in the morning.
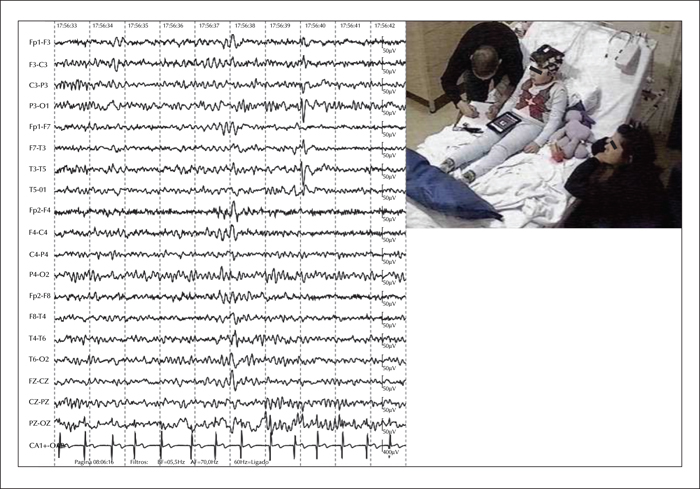
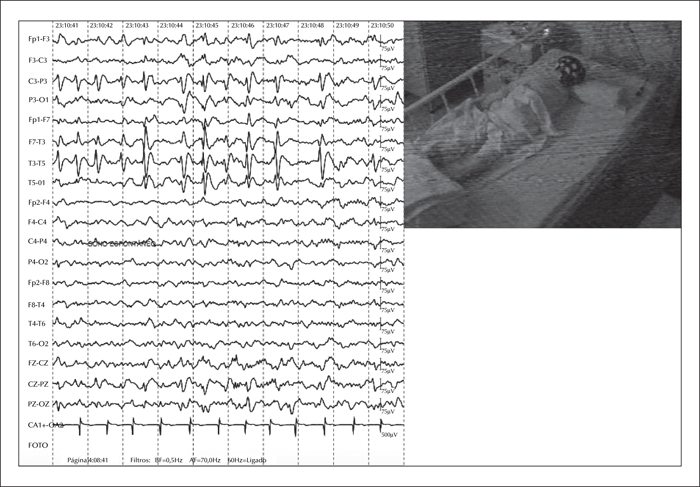
Subsequently, he was admitted to our epilepsy monitoring unit (EMU) for further seizure characterization. During the 72-hour EMU admission, his interictal EEG showed biphasic sharp waves frequently associated with slow waves, generating multiphasic spike-slow wave complexes, emanating from the left Rolandic-temporal region (figure 1). Notably, these complexes had a tendency to spread to adjacent areas and had an increased frequency during NREM Stages 1 and 2 (at times resembling electroencephalographic seizures) (figure 2). Importantly, despite the increase in spike-slow wave complexes during early stages of sleep, the patient's EEG did not meet the classic criteria for electrical status epilepticus during sleep because the discharges occupied less than 40% of the sleep tracing during NR 2-3. Ictally, we observed similar paroxysms that showed progressive increase in frequency with phase reversal at T3, that evolved to an ictal theta rhythm and maintained the same distribution. The clinical events, which lasted a couple of minutes each, featured abrupt awakening, excessive sweating, and prominent fear, which prompted the patient to desperately scream for his mother and subsequently cling to her (video sequence).
Currently, he has been seizure-free for the past year and a half on sulthiamine, 50 mg daily, and valproate, 250 mg daily. He attends regular school with a normal curriculum and does not require tutoring. His intellectual and social abilities are excellent (IQ of 116), although he does have some difficulties particularly in language learning, and was recently diagnosed with phonological dyslexia with dysorthorgraphy. Repeat 3T brain MRI was unremarkable and interictal FDG-PET showed mild hypometabolism in the left lateral temporal lobe. Based on the electroclinical data presented above, we diagnosed this patient with BFEAS.
Discussion
BFEAS, or benign psychomotor epilepsy, is a phenotype of BCSSS, sharing features with Rolandic epilepsy and Panayiotopoulos syndrome (hypersalivation and speech arrest). BFEAS is characterized by paroxysms of sudden fear and screaming, autonomic disturbances (such as sweating, hypersalivation, repeated swallowing, and pallor), automatisms (i.e. chewing), and impaired consciousness. Seizure onset is typically between 2 and 9 years of age. The seizures are brief (lasting a few minutes), can occur both during wakefulness or sleep, and are easily controlled with antiepileptic drugs. As a rule, generalized seizures are not observed in these patients. Postictally, fatigue and/or confusion may occur. Intellectual and social outcomes are usually excellent. Electroencephalographically, interictal EEG shows high-amplitude fronto-temporal and parieto-temporal spikes that are enhanced by sleep, and ictal EEG discharges are localized in the fronto-temporal, centro-temporal or parietal regions and are believed to be stereotypical for each patient (Dalla Bernardina et al., 1992; Panayiotopoulos et al., 2008).
Specifically regarding our patient, a diagnosis of BFEAS was made based on his epilepsy history and seizure semiology (sudden fear and subsequent clinging to his mother, accompanied by autonomic disturbances and impaired consciousness, with seizure onset occurring at 2 years of age), good long-term seizure outcome (seizure-free on valproate and sulthiamine for the past year), electrographic characteristics (spike-slow wave complexes arising from the Rolandic-temporal region that were enhanced by NREM sleep), and excellent intellectual outcome.
It should be noted, however, that some findings in this patient's history are uncommonly observed in BFEAS: the fact that he had multiple episodes of secondary generalized convulsive status epilepticus, and his difficulty in language learning. Nevertheless, both findings have been reported in patients with benign childhood epilepsy with centro-temporal spikes (also known as Rolandic epilepsy) (Kramer et al., 2002; Monjauze et al., 2005; Fejerman, 2009; Ibanez Mico et al., 2012).
An alternative diagnosis would be temporal lobe epilepsy secondary to a slow growing tumour. This condition could explain the initial high seizure frequency (including the multiple episodes of status epilepticus) and early drug resistance followed by well-controlled epilepsy. Although this hypothesis seems to be less likely given our patient's unremarkable initial and repeat brain imaging, careful follow-up is warranted. Other possible differential diagnoses include LGI1-related temporal lobe epilepsy (however, unlikely given that our patient had no auditory features or family history of seizures) (Ottman et al., 2004) and more rare genetic focal epilepsy syndromes, such as those associated with SCN1A, SCN3A, DEPDC5 or PCDH19 mutations (Marini et al., 2012; Vanoye et al., 2014; Striano et al., 2015; McDonald et al., 2017). Lastly, based solely on our patient's clinical presentation (excepting his EEG), pavor nocturnus would also be a possible differential diagnosis.
Diagnosing sudden nocturnal events with affective symptomatology in children is extremely difficult and requires a great amount of knowledge on the main possible differential diagnoses: nightmares, nocturnal panic attacks, pavor nocturnus, psychogenic reactions, and epilepsy (such as autosomal dominant frontal lobe epilepsy and BFEAS). At the same time, diagnostic accuracy is of paramount importance since treatment differs significantly for each aetiology (Cornaggia et al., 2010; Akiyama et al., 2014)
Specifically regarding our patient, a diagnosis of BFEAS was made based on clinical and electrographic characteristics. It should be noted that BFEAS, a phenotype of BCSSS, is characterized by features common to both Rolandic epilepsy and Panayiotopoulos syndrome.
By presenting our patient's history and video-EEG features, which are fairly typical of BFEAS, we provide further detail of the semiology of seizures with affective symptomatology as a reminder to neurologists/epileptologists of this peculiar diagnosis.
Acknowledgements and disclosures
The authors gratefully acknowledge Dr. Guilberto Minguetti and Dr. Vinicius Ludwig for financing and interpreting the PET scan.
None of the authors have any conflict of interest to declare.