Epileptic Disorders
MENUStartle response: epileptic or non-epileptic? The case for "flash" SMA reflex seizures Volume 3, numéro 1, Mars 2001

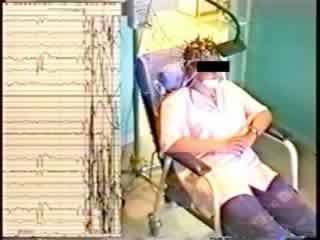
A 19 year-old, female history student attended as an out-patient because of an excessive reaction to sudden, unexpected noise (such as the telephone ringing), as well as to other somatosensory stimuli, such as unexpected taps on the shoulder or unexpected contact involving other parts of her body. She recently had nearly had an accident when driving her car, due to a "jerk" provoked by the horn of another car, which caused her to let go of the steering wheel. She experienced these annoying sudden movements several times a day, often following minor stimuli from the normal environment. She reported that such stimuli caused her to raise her arms, bend forward, lose muscle tone (she sometimes fell), especially in her left leg. All this was very short-lived and she did not lose consciousness.
These episodes had begun in early primary school years and had become much more frequent in recent years. The patient's social life was greatly restricted by these episodes. The patient and her parents confirmed that she never had attacks without provocation. She had never experienced other phenomena suggestive of complex focal or secondarily generalised seizures. No similar case was known in her family. The neurological examination was normal. There was no hemiparesis or pyramidal symptoms. She was intellectually normal, and there were no signs of abnormal behaviour or mood disorder. The magnetic resonance imaging (MRI) scan was normal. The baseline EEG recording was normal. The clinical phenomenon was easily produced during the interview and during the EEG recording, by an unexpected loud noise (see video). On that particular day, tapping her forehead was not effective in inducing a startle response, and "expected" stimuli (noise, contact) were also ineffective. The ictal EEG polygraphy clearly demonstrated the startle reaction, with a diffuse muscular contraction over both deltoid muscles. The ictal EEG was difficult to read due to movement artefacts (figure 1). A short run of low-voltage, fast activity may have occurred over the central area approximately one second after the stimulus, followed by some slow waves over C4. We did not consider the EEG changes to be sufficient by themselves for a definite diagnosis. However, there was a significant past history.
She had been born at term to healthy parents after an uneventful pregnancy and delivery. At two months of age, she experienced a series of apparently generalised tonic-clonic convulsions, without fever and was thereafter repeatedly admitted to a general hospital. An interictal waking and sleep EEG showed spike and wave activity in the right frontal region as well as generalised discharges. The seizures were controlled by phenobarbital (PB) and diazepam within 24 hours. Neurological examination was normal and cranial computerised tomography (CT) was normal. While still on PB, at three months old she experienced brief episodes of sudden, diffuse contraction often but apparently not always triggered by noise. These data were obtained from her parents and from the early medical records, which were recovered from the medical files of a local hospital, together with samples from the early EEGs. The attacks occurred mostly during drowsiness or awakening, although not in clusters or series, and a diagnosis of infantile spasms was made based on persisting abnormal EEG findings. The original tracings (figure 2a) showed diffuse sharp abnormalities more evident over the anterior regions, predomination on the right, especially during sleep, and an isolated extensor spasm was reportedly recorded (figure 2b). Treatment with ACTH was initiated and the clinical phenomena completely disappeared within 10 days, and did not recur. Developmental milestones were normal but she was first able to walk alone at 16 months of age. PB was maintained until four years of age. Her parents did not link the startles that appeared at around age six to the former condition and never sought medical advice.
We thus considered that this patient had a reflex epilepsy, with very early onset, apparent remission in the first year of life following ACTH therapy, and relapse six years later. We started therapy with carbamazepine (CBZ), at 200 mg/d; there was no improvement: at 400 mg/day, there was a marked reduction in the frequency of startles, and a complete resolution of symptoms was obtained at 600 mg/d (blood levels: 7.9 mg/l). She has remained free of symptoms for two years, except for periods when she decided to reduce her medication or even to stop it altogether. She then experienced a recurrence of the startles.
An excessive startle reaction may occur in various conditions. It is a feature of startle epilepsy and of non-epileptic disorders such as hyperekplexia and jumping [1-3] or even La Tourette's syndrome [4]. Hyperekplexia or startle disease is characterised by an excessive startle response to tactile, auditory or visual stimuli which often results in a precipitous fall (review in [3]). Consciousness is preserved during the episodes. It is usually an autosomal dominant hereditary disorder [5], but autosomal recessive transmission [6] and sporadic occurrence have been reported [1]. Hyperekplexia was also shown to be linked to a mutation in the gene coding for the alpha1 subunit of the glycine receptor, on the short arm of chromosome five [6, 7]. The disorder occurs in two forms: in addition to exaggerated startle response, the major form may start in the neonatal period, manifesting as generalised muscular hypertonia in infancy which disappears during sleep, and by nocturnal myoclonus. Touching the dorsum or the tip of the nose elicits symmetric jerks of all four limbs with no habituation, and a tonic attack with apnea may occur. This fit can also occur during sleep. There is a progressive improvement of rigidity and of attacks with age, but exaggerated startle response persists into adulthood. The minor form is characterised by startle response only without congenital hypertonia or prominent nocturnal myoclonus. Although some patients may experience infrequent epileptic seizures, hyperekplexia is not associated in the infant with important interictal paroxysmal changes [3]. In the first year of life, hyperekplexia is often confused with epilepsy.
Startle epilepsy (SE) is characterised by seizures induced by sudden and unexpected stimuli and usually occurs in patients with cerebral lesions, most of which originate during the prenatal or perinatal periods [8]. The seizures are frequent, usually lasting less than 30 seconds, and consist of a startle response followed by a brief but characteristic tonic phase which is usually asymmetric. Spontaneous seizures reportedly occur in all cases, but may be infrequent and escape attention. Spontaneous seizures typically precede startle-induced seizures by years. SE has been reported in patients with cerebral palsy, perinatal anoxia [8], Lennox-Gastaut syndrome [9], Sturge-Weber syndrome [10], Tay-Sachs' disease [11], Reye's syndrome [11], epidermal naevus syndrome [12], and Down syndrome [13]. Patients with SE usually have evidence of localised or diffuse static encephalopathy or developmental delay. Neuroimaging may show localised or diffuse lesions but normal results have been reported without neurological deficit. Aguglia et al. (1984) [11] reported clinical and EEG data in 16 cases with startle epilepsy: ten had congenital brain damage, while five had acquired encephalopathy or encephalitis and only one patient was neurologically normal with a normal CT scan. However, the detection of lesions may depend on the quality of neuroimaging: in four patients with normal CT or conventional MRI scans and no neurological deficit, high-resolution MRI showed a focal migration disorder in three (perisylvian, 1, frontal, 2) and only one was normal [14]; however, the latter patient had interictal bifrontal spikes on the EEG.
The primary epileptogenic zone in SE has been described as the primary motor cortex, the premotor cortex and the supplementary motor area (SMA), using depth electrodes [15-17] or subdural grids [18]. According to these authors, typical features of SMA seizures include asymmetric tonic posturing of the extremities, abduction and elevation of the contralateral upper extremity, and speech arrest with preserved consciousness. Our patient never experienced other types of ictal events besides the very brief, strictly reflex phenomena she referred to as "jerks". The ictal symptomatology was very brief but the sequence of motor events (see video) was fully compatible with the classical descriptions of SMA seizures. The events were however, too brief for speech arrest to become prominent, and there was no vocalisation. Furthermore, we could only provide surface EEG recordings, as the condition proved to be benign, and the tracing failed to show a clear-cut ictal activity, althought some elements did favour the diagnosis of an epileptic event (figure 1).
Many elements were in favour of SE. Our patient demonstrated no evidence of hypertonia or generalised muscular rigidity during infancy. Prominent startle responses, without falls were noted at an early age and recurred in childhood after several years of apparent remission. There were marked EEG changes in early childhood. The clinical symptoms appeared as very stable over the years, since they relapsed around the age of six, and the untreated condition worsened steadily through childhood and adolescence instead of remitting. There was a quick and full response to anticonvulsant treatment using CBZ. On the other hand, some clinical elements did favour the diagnosis of hyperekplexia, which may occur sporadically: very early age at onset, no other associated seizure beyond infancy, no spontaneous seizures, slight motor retardation, with walking at age 16 months only. However, the absence of abnormal neuroimaging remains compatible with the diagnosis of SE, as shown above; it must be stressed that the patient did not have high-resolution MRI, which might have shown a discrete cortical lesion.
First part (repeated once): at normal speed. A sudden, violent and unexpected noise provokes a bilateral abduction of the upper limbs. On the left, the arm is raised horizontally and flexed. The right hand grasps the arm of the chair. The trunk and head are bent forward, and she closes her eyes. The lower limbs are abducted, and the right thigh is slightly raised. There is no loss of contact during this very brief event.
Second part: slow motion, in order to detail the sequence of motor events.
CONCLUSION
This patient had features suggestive of hyperekplexia and SE. Using past history, and clinical and EEG symptomatology, we concluded that she had an unusual form of reflex, "startle" epilepsy; i.e. a cryptogenic focal epilepsy with exclusively reflex seizures. Seizures were particular in the sense that they represented very short ictal events, which were probably located in the SMA. We propose to use the term "flash" to describe such an unusual, very brief event, in order to distinguish it from the longer, tonic seizures ususally recorded in this setting, and to differentiate it from myoclonic events. We chose to treat her with CBZ, a first-line drug for focal epilepsies. The clinical response was excellent and clearly dose-related. Does this fully validate our diagnosis? Similar observations may have been documented elsewhere, and the authors would be very interested in discussing them
Received August 21, 2000 / Accepted January 15, 2001