Epileptic Disorders
MENUSotos syndrome: a pitfall in the presurgical workup of temporal lobe epilepsy Volume 23, numéro 3, June 2021

Illustrations
Epilepsy surgery is a very effective treatment for selected children and adolescents with drug-resistant focal lesional epilepsy [1]. A quarter of paediatric patients undergoing epilepsy surgery have a tumour-associated epilepsy [2] and three quarters of these patients achieve long-term seizure freedom, with the majority being able to stop anti-seizure medication [3]. Successful surgery for tumour-associated epilepsy in early life can lead to cessation of prolonged or even life-threatening seizures, continuous epileptic discharges, polytherapy, and epilepsy-related developmental arrest or regression that severely impact the quality of life for both the affected child and their family [3]. Recently, the view that surgery for tumour-associated epilepsy should be offered before the criteria of pharmacoresistance are met has been rapidly gaining ground [4].
The genomic revolution of the last decades has produced a growing body of evidence pointing to an underlying genetic cause for many patients with focal or generalized epilepsies [5]. These include single gene mutations in genes related to ion channel function, synaptic transmission, or the mammalian target of rapamycin (mTOR) pathway, as well as several recurrent microdeletions and other chromosomal abnormalities [6]. With increasing access to genomic diagnostics, patients undergoing evaluation for epilepsy surgery are now sometimes also tested for genetic causes for their epilepsy, which, in the absence of MRI-visible structural lesions or mTOR-pathway mutations, tends to be associated with a worse post-surgical seizure outcome [7]. Despite these novel insights, genetic workup is not yet a standard component of presurgical evaluation.
Case study
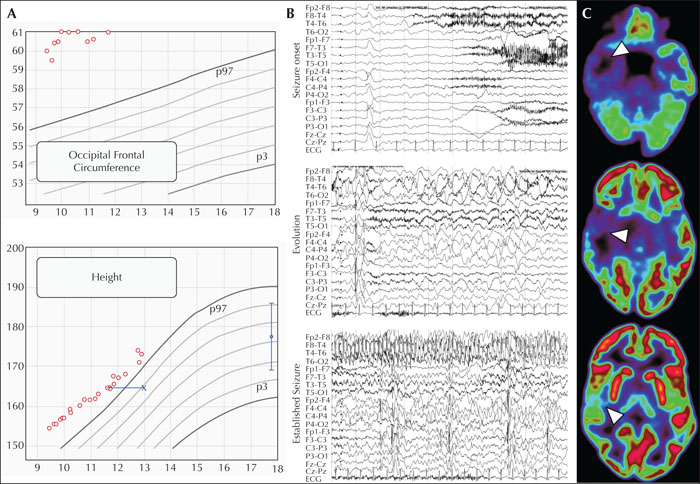
A six-year-old boy, born after uncomplicated pregnancy to unrelated parents, with high anthropometric measures at birth (>97th centile), experienced his first unprovoked focal seizures. These occurred in wakefulness and were characterized by behavioural arrest, staring, and oroalimentary and manual automatisms, sometimes preceded by an unpleasant abdominal sensation, with evolution to bilateral tonic-clonic seizures within a year from seizure onset. Interestingly, the boy sometimes described seeing “headless figures” and had intense emotional outbursts related to his seizures, e.g., crying and claiming: “I wish I was dead”. Past medical history was significant only for prior tonsillectomy and tympanostomy tube insertion in the first year of life, with persisting obstructive sleep apnoea into later childhood. Development according to the parents was unremarkable prior to first seizures, even though retrospectively it became apparent that emotional dysregulation, attentional deficits and problems with social interaction preceded his epilepsy onset. His family history was unremarkable. Examination at presentation showed a large occipital frontal circumference (>97th centile) and tall stature (>97th centile) but endocrinological investigations were normal.
Repeat interictal EEGs showed continuous right (fronto-) temporal slowing with superimposed sharp waves as well as abundant diffuse bilateral sharp waves and polyspikes, at times with frontal to fronto-temporal predominance. Brain MRI revealed a right temporal lesion affecting the mesial temporal structures, the inferior temporal gyrus, and the parahippocampal gyrus, suggestive of an astrocytoma or ganglioglioma (figure 1A). The general clinical impression was that of a focal-onset epilepsy associated with a low-grade neuroepithelial tumour. Seizures remained refractory to treatment, first with oxcarbazepine as monotherapy (maximum daily dose of 42 mg/kg), and later in combination with levetiracetam (maximum daily dose of 60 mg/kg). At this point, the patient was referred for evaluation for epilepsy surgery.
He underwent video-telemetry that captured several seizures following the tapering of his anti-seizure medication. These occurred out of sleep upon awakening and with blinking, restlessness, unresponsiveness, head/upper body deviation to the left, right manual automatisms, oroalimentary automatisms, vocalization, and tonic posturing of the left arm. Seizures lasted up to 2.5 minutes, followed by rapid reorientation with intact language function, and then postictal sleep. Ictal EEG (figure 1B) showed right temporal high-amplitude rhythmic slowing with evolution from theta- to delta-band activity, transition to repetitive high-amplitude sharp slow waves with right mid-temporal onset, and subsequent maximum and propagation over the right hemisphere, followed by right temporal postictal slowing. In addition to fragmented sleep architecture, frequent, presumably epileptic, arousals were recorded that evolved from the interictal right (fronto-) temporal slowing. In line with the EEG and MRI findings, FDG-PET showed pronounced right temporal hypometabolism (figure 1A), supporting the hypothesis of a right temporal epileptogenic zone, matching the location of the tumour. Neuropsychological testing before surgery showed overall cognitive functions slightly below the average range with problems in logical thinking and working memory, reduced selective attention, reduced verbal drive, significant visuo-constructive problems and considerable bimanual fine motor deficits.
In light of the overall congruent semiology, interictal and ictal EEG, MRI and PET findings compatible with a right mesial temporal epilepsy, the patient was offered surgical resection of his tumour, despite the abundant diffuse bilateral sharp waves and polyspikes on interictal EEG and the pre-existing developmental delay and behavioural issues not fully explained by his epilepsy. The surgery was performed at the age of 10 years. Histopathology revealed a diffuse astrocytoma.
Three months after surgery, EEG showed persisting bilateral diffuse sharp slow waves and polyspikes, while MRI showed gross total tumour resection. Six months after surgery, the patient presented with seizure recurrence out of night sleep, with tonic posturing in his arm and leg, albeit with unclear lateralization, followed by prolonged and inconsolable crying. Over the following two years, anti-seizure medication was changed to lacosamide with later addition of perampanel. EEG findings remained unchanged, with a persistence of presumably epileptic arousals with a temporal EEG pattern, sometimes linked to the postsurgical right temporal slowing, and recurrent focal seizures with temporal lobe semiology every few months, albeit with inconstant lateralizing signs. In addition, the patient had increasing emotional and behavioural problems and frequent mood swings.
Due to the poor response to anti-seizure medication, the unusual postsurgical course with seizure recurrence with a temporal lobe semiology, albeit with inconstant lateralizing signs, the diffuse EEG findings in the absence of a residual MRI-visible lesion, the evolving, now prominent behavioural and emotional issues, and an increasingly apparent unusual facial appearance with a remarkably tall stature, we initiated trio whole-exome sequencing. The diagnosis of Sotos syndrome, considered due to the overgrowth features of the patient, was confirmed by the detection of a likely pathogenic variant in the NSD1 gene. The diagnosis of a genetic epilepsy commonly presenting with temporal lobe seizures in the absence of a remaining structural lesion deterred us from further presurgical investigations.
Discussion
We report the first case of (failed) epilepsy surgery in a patient with Sotos syndrome, drug-resistant temporal lobe seizures, and a diffuse astrocytoma in the temporal lobe. Epilepsy surgery with gross total resection of the structural lesion failed to achieve seizure control due to the underlying genetic aetiology of Sotos syndrome which is often linked to the electroclinical features of temporal lobe epilepsy in the absence of a brain lesion.
Sotos syndrome (OMIM #117550) was first described in 1964 by Dr Juan Sotos [8] as an overgrowth-intellectual disability (OGID) syndrome. The cardinal clinical features of Sotos syndrome include a characteristic facial appearance (broad and prominent forehead and a dolichocephalic head shape, sparse frontotemporal hair, downward-slanting palpebral fissures, malar flushing, long and narrow face, and a long chin), learning disability (early developmental delay and, eventually, mild-to-severe intellectual impairment), and overgrowth, resulting in tall stature and macrocephaly. Major features include neonatal complications, bone age, scoliosis, joint hyperlaxity, renal and cardiac anomalies, behavioural and emotional problems, and seizures [9]. In addition, otolaryngologic problems may occur such as conductive or sensorineural hearing loss, aspiration, laryngomalacia, and obstructive sleep apnoea [10]. Sotos syndrome is caused by pathogenic loss-of-function variants, or deletions encompassing the nuclear receptor set domain containing protein 1 gene (NSD1), located at 5q35.2–q35.3 [11]. It is inherited in an autosomal dominant manner but more than 95% of individuals have a de novo pathogenic variant or demonstrate copy number variations [9].
Sotos syndrome is associated with increased incidence of malignancy with a 2-3% risk of developing neoplasms [12]. Lympho-haematological tumours are most frequently encountered, with non-Hodgkin lymphoma comprising almost a quarter of reported cases. Other types of neoplasms include acute lymphoblastic leukaemia, hepatoblastoma, Wilms’ tumour, gastric carcinoma and small cell lung cancer, while brain neoplasms have only rarely been reported [13, 14]. Brain MRI findings in patients with Sotos syndrome suggest delayed or disturbed maturation, particularly of midline structures, and include ventricular anomalies, enlargement of supratentorial extracerebral fluid spaces, and anomalies of the corpus callosum and other midline structures [9, 15]. So far, low-grade tumour associated with epilepsy of Sotos syndrome has been reported in only two cases; in a child and an adult with low-grade glioma [13] and astrocytoma [14].
Seizures occur in 15-50% of patients with Sotos syndrome, are initially associated with a febrile illness in half of the cases, and include diverse semiologies such as epileptic spasms, absences, and focal seizures with or without evolution to bilateral tonic-clonic seizures [9, 16]. Seizures usually respond well to anti-seizure medication, and they wane over time [15] and only seldom persist (9%) into adulthood [17]. Drug resistance is rare and has so far only been reported in relation to periventricular nodular heterotopia [15, 18]. Reportedly 40% of patients with Sotos syndrome have temporal lobe seizures characterized by abdominal déjà vu or jamais vu auras, fear, and automatisms with/without behavioural arrest and a temporal onset on ictal EEG, albeit in the absence of associated brain MRI abnormalities [15]. Temporal lobe seizures may include pronounced behavioural elements, such as aggressive or emotional outbursts, that hamper their differential diagnosis that includes aggravation of the behavioural disorder. Tonic-clonic seizures occur in a third of patients with Sotos syndrome, often evolving from focal seizures. Notably, focal slowing and epileptic discharges over the temporal regions, matching the temporal lobe semiology, are common findings on interictal EEG of patients with Sotos syndrome [15].
Our case highlights Sotos syndrome as a rare but important pitfall in the presurgical workup of temporal lobe epilepsy that should be considered particularly in MRI-negative cases but also in the presence of a focal lesion that does not fully explain the clinical picture. Most importantly, our observations underline the value of thorough presurgical diagnostics, including genetic testing. In our case, the pre-existing developmental delay and behavioural issues in the context of medical comorbidities could have prompted an earlier genetic work-up. Extensive genetic testing may help rule out an aetiology that can potentially limit the chances of postoperative seizure freedom. Finally, our case emphasizes the need to re-evaluate our less successful epilepsy surgery cases in order to offer informed counselling and prognostication, even if further surgery is not an option [19].
Supplementary material
Summary slides accompanying the manuscript are available at www.epilepticdisorders.com.
Disclosures
None of the authors have any conflict of interest to declare.