Epileptic Disorders
MENUKCTD7-related progressive myoclonus epilepsy Volume 18, supplément 2, September 2016

- Mots-clés : KCTD7, progressive myoclonus epilepsies, infancy, encephalopathy
- DOI : 10.1684/epd.2016.0856
- Page(s) : 115-9
- Année de parution : 2016
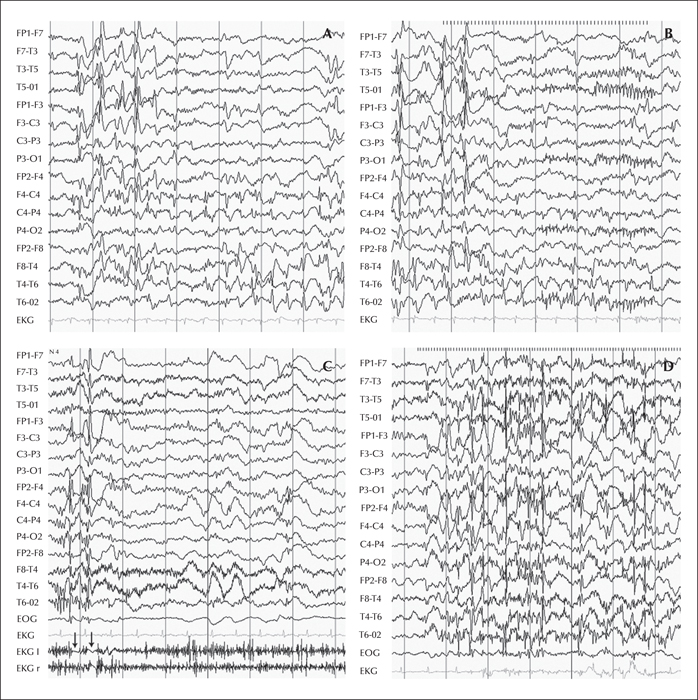
Progressive myoclonic epilepsy associated with KCTD7 mutations has been reported in 19 patients from 12 families. Patients show homozygous mutations in the coding regions of the KCTD7 gene. The disease starts in infancy. Patients typically show an initial severe epileptic disorder, with abundant epileptiform discharges on EEG and myoclonic seizures in the foreground, associated with cognitive regression and ataxia. Continuous multifocal myoclonus aggravated by action is observed in more than half of the cases. After a few years, the disease tends to stabilize and long survival can be expected. Some patients remain able to walk independently. The severity of the disease is variable from one patient to another, even within the same family. It is hypothesized that the epileptic disorder may influence the neurological regression observed in patients.