Epileptic Disorders
MENUHow to diagnose and classify idiopathic (genetic) generalized epilepsies Volume 22, numéro 4, August 2020

Illustrations



Tableaux
Diagnosis of epilepsy is essentially clinical, based on skilful history taking and physical and neurological examination. Once paroxysmal imitators of epileptic seizures, such as reflex convulsive syncope and psychogenic non-epileptic seizures are ruled out, diagnosis of the clinical epilepsy type and syndrome, and identification of the underlying cause are mandatory for appropriate management and treatment, also considering the individual patient's circumstances, including comorbidities and needs.
The new ILAE classification of the epilepsies (Scheffer et al., 2017) provides an orderly diagnostic process through three steps (levels) of increasing complexity to adapt to the available resources. The first step is the diagnosis of seizure type (of focal, generalized or unknown onset), according to the recent diagnostic criteria (Fisher et al., 2017).The second step is the diagnosis of the epilepsy type (focal, generalized, combined focal and generalized and unknown), according to the seizure or combination of seizures manifested. The third level is the diagnosis of the particular epilepsy syndrome, when possible. Identification of the underlying cause should be pursued as soon as possible (Scheffer et al., 2017).
As in any other disorder, the first step of the diagnostic process begins with a thorough medical history and full physical / neurological examination. History taking should strive to obtain accurate information about seizure symptoms and semiology from the patient and possible observers, as well as about seizure timing and circadian distribution, circumstances, facilitators and possible triggers, antecedents for seizures, comorbidities and relevant family history. The available information (on the basis of which imitators of epilepsy can also be ruled out) leads to the formation of a provisional diagnostic (or differential diagnostic) hypothesis, for the testing of which appropriate diagnostics are requested, including brain imaging, EEG, blood and urine tests, etc. Unless the clinical/EEG picture indicates an idiopathic focal or generalized epilepsy, recognition of aetiology typically requires brain imaging and other laboratory investigations. Synthesis of the clinical and laboratory evidence will lead to strengthening (or revision) of the provisional hypothesis into a definite or highly probable diagnosis, on which initial treatment and management are based. It has to be emphasized that this “final” diagnosis should not permanently be set in stone but may remain subject to revisions in case other information comes to light or a new seizure type is further added during the natural course of the epilepsy. Indeed, the ability to revise a diagnosis is an important virtue of the competent open-minded physician.
In this paper, we first discuss the clinical presentation of the main seizure types that are encountered in patients with idiopathic generalised epilepsy (IGE), focusing on clinical diagnostic clues on the basis of which the initial (working) diagnostic hypothesis can be made. We then refer to the basic principles and role of the EEG in the diagnosis of IGE, delineating its uses and limitations. This paper concludes with a brief discussion about the main IGE syndromes, emphasising the relevant diagnostic features. For more detailed description of the interictal and ictal EEG findings in IGE and its various syndromes, readers are referred to the comprehensive educational paper, published in this journal (Koutroumanidis et al., 2017). This educational review paper addresses the following learning objective of the ILAE curriculum in epileptology: Correctly diagnose and classify generalized epilepsies (Blümcke et al., 2019).
When seeing a patient with probable IGE...
The first thing to do is to acquire a complete history in order to fully and clearly comprehend the clinical picture. The type or types of seizure, age at their onset, and their frequency, timing and triggers should all be meticulously sought. A detailed description of each seizure type should be obtained from the patient and witnesses. Asking too many open-ended questions may distract the patient, whereas asking only yes/no questions may create a suggestion bias.
The clinically prominent seizure type and the possible existence of multiple seizure types are a good starting point for a working IGE differential diagnosis. Prominent myoclonic seizures (MS) combined (or not) with typical absences (TA) and rare generalized tonic-clonic seizures (GTCS) suggest juvenile myoclonic epilepsy (JME), whereas a patient presenting with prominent TA is more likely to have childhood (CAE) or juvenile absence epilepsy (JAE). GTCS without concomitant MS or TA would suggest a diagnosis of IGE with GTCS alone (GTCS-a). Age at onset is another important issue in the diagnostic process. Typical absences starting well before the age of 10 years suggest a diagnosis of CAE, whereas patients with seizures starting around 10 years of age or later are more likely to have JAE or JME. The timing of seizure occurrence can aid the clinician in the diagnostic process since some of the IGE subgroups show strong circadian characteristics. Myoclonic jerks (MJ) in the morning is almost a clinical hallmark of JME, while GTCS also tend to occur upon awakening in the majority of patients.
Another important point is the absence of complications based on the history and examination, other than seizures. Additional neurological or cognitive signs and symptoms are not expected in IGE patients and should be interpreted as “red flag” findings. Mild behavioural problems or personality traits can be expected in some patients, especially those with JME, while sometimes, patients with frequent absences may have been misdiagnosed with cognitive problems due to inattention during their clusters of absences; however, any other prominent sign or symptom should warrant additional diagnostic consideration.
Family history can be instructive in certain cases, as it is quite common for other family members to have IGE. A mother with morning jerks or a first uncle who had “vacant spells” when he was young, “but subsequently was ok”, can be a valuable hint for diagnosing these patients.
If the clinician is at ease with the IGE diagnosis, EEG evidence is supportive, and there are no red flag signs suggesting otherwise, structural imaging is not a necessary part of the diagnostic workup for IGE. If performed, it is expected to be normal unless either the initial diagnosis is erroneous, or a comorbid non-causal disorder exists.
EEG is an indispensable tool in the diagnostic process. While normal routine EEG in IGE patients is not unexpected, the sensitivity of the EEG can be increased via many methods. Sleep deprivation and sleep can enhance generalised spike-wave discharges (GSWDs), the EEG hallmark of IGE. Hyperventilation is the foremost method triggering GSWDs and TA, and can be performed multiple times during the same recording, as required. Photic stimulation can reveal photosensitivity and other historical triggers can be tested under controlled and safe conditions. Scheduling the EEG appointment to after awakening may disclose GSWDs and MS which could have been otherwise missed in a JME patient. Twenty-four-hour EEG or longer telemetry may allow registration of GSWD when routine or sleep EEG is unremarkable, although such recordings are rarely needed. Therefore, clinicians should be precise about the objective of the EEG and know what they are looking for in order to find it.
On the concept of IGEs/GGEs: from seizures to syndromes
Generalised epilepsies manifest with generalised seizures, and amongst these, idiopathic/genetic epilepsies are the most prominent group that comprises almost a third of all epilepsies. The term “idiopathic” comes from the Greek words “idios” that means “oneself” and “pathos” that means “suffering”, and conveys the notion of “only epilepsy”, without any causal brain lesion or other genetic demonstrable aetiology, hence the complementary term “genetic generalised epilepsies”. Of note, “idiopathic” is not synonymous with “benign”. Indeed, IGE is long-term pharmaco-resistant in about 20% of patients (Vorderwülbecke et al., 2017). There are three main seizure types in IGE, namely generalized tonic-clonic seizures (GTCS), typical absences (TA) and myoclonic seizures (MS), occurring either alone or in any combination. Absence status can also be considered as a distinctive seizure type. Generalised tonic seizures occur only exceptionally. The age at onset and the seizure type or combination of seizure types are the main clinical criteria for the classification into IGE syndromes. There exist four well-established syndromes and some other rarer phenotypes. The main four IGE syndromes are childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), juvenile myoclonic epilepsy (JME) and IGE with generalized tonic-clonic seizures alone (GTCS-a).
Definition of generalized-onset seizures
According to the 2010 ILAE report on revised terminology and concepts for organization of seizures and epilepsies (Berg et al., 2010), “generalized epileptic seizures are conceptualized as originating at some point within, and rapidly engaging, bilaterally distributed networks”. Although this concept presupposes that in these seizures the first ictal clinical changes reflect involvement of both hemispheres and the initial EEG changes are bilateral and apparently synchronous, as we shall see below this is not always the case. Thus, the ILAE definition duly goes on to clarify that “although individual seizure onsets can appear localized, the location and lateralization are not consistent from one seizure to another. Generalized seizures can be asymmetric”.
In the next section, each of the main IGE seizure types is clinically described along with practical diagnostic clues. It is reminded that the initial diagnostic hypothesis must be corroborated by concordant EEG findings. For instance, a clinically convincing history of generalized convulsions without supportive EEG evidence (i.e. with normal EEG) is not enough to make the diagnosis of GTCS-a (Scheffer et al., 2017).
Generalized tonic-clonic seizure (GTCS)
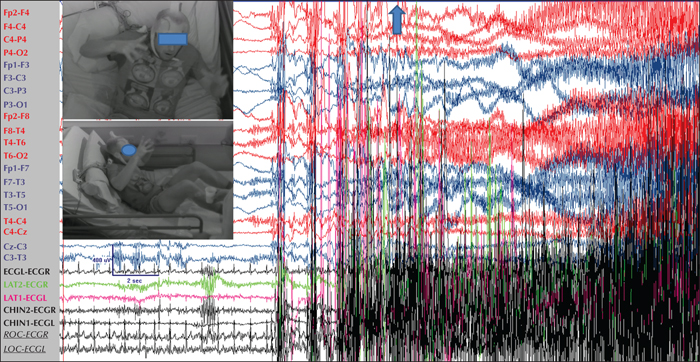
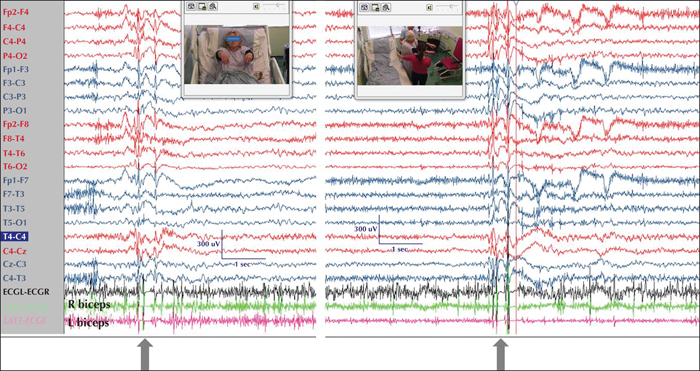
True to their former name “grand mal”, GTCS are terrifying for the patients and their relatives. They tend to last between 40-70 seconds but sometimes may be longer (Nordli et al., 2011). Onset is sudden; eyes are open (and remain open thereafter throughout the seizure) and consciousness is instantly and completely abolished until the postictal recovery period when it is gradually restored. In some patients with JME, the onset of the actual GTCS may immediately follow volleys of bilateral myoclonic jerks in a “myoclonic-tonic-clonic sequence”, while in some patients with JAE or another IGE syndrome with absences, the onset may follow a typical absence or absence status (“absence-tonic-clonic sequence”).The tonic phase is characterised by a sustained contraction of all body muscles, first in flexion (figure 1) and then in extension (figure 2). The tonic contraction of the respiratory muscles results in cyanosis and also in the so-called “epileptic cry”, a high-pitched loud scream produced by air being forced through the closed vocal cords.
The tonic phase lasts about 10 to 20 seconds and transcends into the clonic phase through an intermediate vibratory stage, during which fast clonic contractions at 8 Hz, slowing down to 4 Hz, are superimposed on the tonic activity. The ensuing clonic phase is characterised by successive massive bilateral clonic spasms that progressively decrease in frequency and amplitude until they stop. If the tongue is bitten, this usually happens in the clonic phase, while enuresis, and rarely also encopresis, occur at the end of this phase (Conradsen et al., 2013).
From onset and throughout the seizure, intense autonomic activation results in dilation of the pupils, increased heart rate and blood pressure, profound hypersalivation and sweating.
After the final clonic jerk, the patient lies comatose and flaccid. Respiration returns initially with stertorous breathing and gradually muscle tone and autonomic function normalise. Awareness also returns gradually through a period of diminishing postictal confusion, which is longer when the GTCS is the first or one of the first of a new epilepsy, or after serial GTCS or convulsive status epilepticus. Alternatively, the patient may slip into a deep postictal sleep, sometimes lasting for hours. Diffuse muscle aches may last for longer.
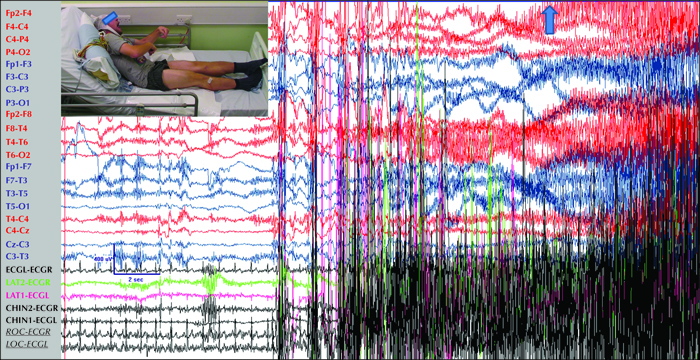
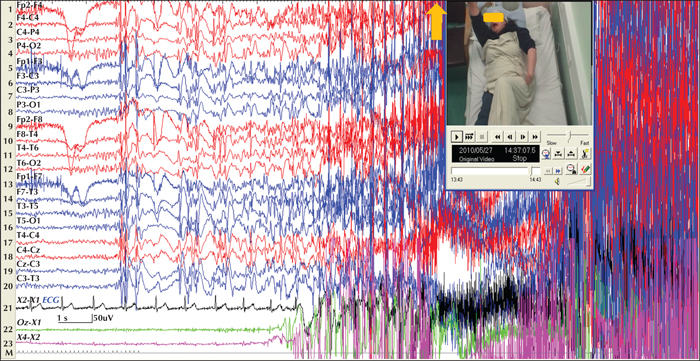
Note: As mentioned above, lateralised or localised initial ictal semiology in true GGE/IGE patients is not uncommon (figure 3). In such cases, “focal” features are usually short-lived and different from a lengthy focal seizure that becomes bilateral tonic-clonic. The reverse may also occur; rapidly generalized focal seizures may lead to a false diagnosis of generalized epilepsy (figure 4).
Practical clinical diagnostic clues for generalised tonic-clonic seizures
GTCS in IGE (as well as typical absences and MS) are, by definition, deprived of any warning, known also as aura; a focal seizure with retained awareness. However, accompanying sensations evocative of auras, and even true auras may be infrequently described by some patients posing diagnostic challenges even for experienced epileptologists (Gungor-Tuncer et al., 2012).One has to firmly keep in mind that a lack of aura does not necessarily mean generalised onset. Focal seizures may become rapidly generalised without initial focal symptoms (as for instance in frontal lobe epilepsy) mimicking GTCS (see also figure 4); or again, they may begin with impairment of awareness (as for instance in limbic temporal lobe seizures) and manifest with psychomotor arrest until they become bilateral tonic-clonic; in the latter case, seizure phenomenology is not different to that of an absence to GTCS sequence.
Despite these difficulties, some clinical clues can be useful to construct the diagnostic working hypothesis. In the case of a generalised convulsion, an account of bilateral myoclonic jerks just before the onset of the diffuse shaking, given either by the patient (as awareness during MS is not disturbed) or by witnesses, would suggest GTCS (myoclonic-tonic-clonic sequence), while a clearly focal seizure immediately preceding the onset would suggest focal to bilateral TCS. A simple “focal” onset, such as brief head turning, may not be enough to clinically distinguish a GTCS from a focal to bilateral tonic-clonic seizure, as brief “focal” symptoms or signs may occur in a sizable proportion of patients with IGE (figure 3; see also Usui et al. [2005] and Gungor-Tuncer et al. [2012]). Of note, family members and eyewitnesses are often too terrified by the experience of seeing a loved one suffer a generalised convulsion to accurately or reliably describe the ictal signs and their order.
Table 1 summarises the main clinical criteria that differentiate GTCS from focal to bilateral tonic-clonic seizures.
Typical absence seizures (TA)
Beautifully named by Calmeil in 1824 (Calmeil, 1824), TA are characterized clinically by sudden impairment of consciousness (absence) that occurs without warning and ceases also suddenly without postictal symptoms, and electrographically by generalized 3-4-Hz spike-and-slow-wave discharge that terminates without subsequent electrical flattening. TA generally last between 2-3 to 20+ seconds. The patient suddenly stops any ongoing activity and appears to stare blankly, eyes may drift upward, and eyelids may flicker, rhythmically or randomly.
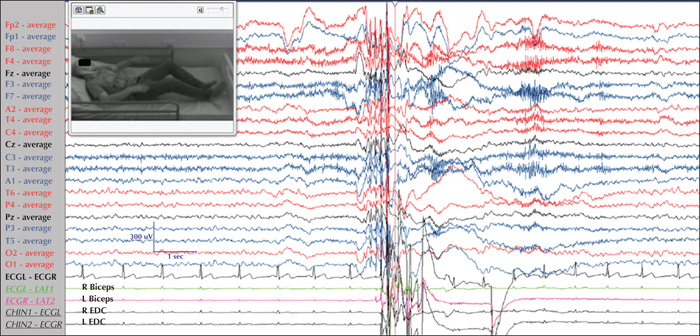
The term “typical” distinguishes these absences from the “atypical” absences that occur in non-idiopathic generalised epilepsies and are associated with subtle impairment of consciousness and slower (Penry et al., 1975; Panayiotopoulos et al., 1989; Capovilla et al., 2001). Although most clinicians tend to prioritize a temporal lobe epilepsy diagnosis in the face of accompanying automatisms, patients with typical absences not infrequently display ictal perioral, limb and speech automatisms (Penry and Dreifuss, 1969; Kessler et al., 2017). Usually, longer absences are associated with more colourful automatisms (figure 5).
Ictal impairment of consciousness varies considerably between patients and even between seizures of the same patient. Typically, it is severe in patients with CAE and JAE, and milder in JME. Phantom absences, may be unperceivably mild, escaping the attention of both the patient and family members or other observers, and may occur in all IGE syndromes. However, if they are the only type of absence, they can delay the diagnosis of IGE until a GTCS occurs and an EEG is performed (Koutroumanidis et al., 2008). Impairment of awareness in such mild and brief absences can be demonstrated by repetitive tasks such as breath counting during hyperventilation or sequential tapping (figure 6). Atypical and deviant features, such as accompanying focal behavioural changes and partially retained awareness, may further complicate the diagnostic process and cause a delay in the correct diagnosis.
Absences with eyelid myoclonia is another peculiar entity. They occur on eye closure and are characterized by myoclonia of the eyelids and associated eye-closure EEG paroxysms. These absences are the defining seizure type of Jeavons syndrome (Striano et al., 2009). They are generally mild, but because eye-closure EEG paroxysms may also be subclinical, Jeavons syndrome is also known as “eyelid myoclonia with or without absences”. Absences associated with rhythmical 3-Hz myoclonic jerks of the head or axial muscles, time-locked to the spike component of the generalised spike-wave discharge, are rare in IGE, whereas they are the distinguishing clinical feature of myoclonic absence epilepsy (MAE). In MAE, myoclonic absence seizures are characterized by rhythmic 3-Hz myoclonic jerks that involve the face and the upper limbs, associated with a tonic contraction involving predominantly the shoulder muscles, causing elevation of the arms. The myoclonic jerks are time-locked to the 3-Hz generalized spike-wave complex (Tassinari et al., 1969; Rubboli et al., 2019). According to the 1989 classification proposal (Commission on Classification and Terminology of the ILAE, 1989), MAE is classified among the “generalized cryptogenic or symptomatic epilepsies”. Indeed, the prognosis of MAE may not be as favourable as that in most cases of IGE.
Practical clinical diagnostic clues for typical absences
TA are easy to appreciate when the presentation is typical for CAE (pyknolepsy, typical circadian occurrence and easy provocation by simple hyperventilation as part of the clinical examination). Absences of later onset, as in JAE or in early adulthood, may be difficult to differentiate from mild focal limbic (temporal lobe) seizures, which may not be followed by obvious postictal symptoms. Brief or mild absences associated only with fleeting impairment of awareness may pass unnoticed by patients and relatives and be denied on direct questioning.
Clinical clues that help differentiate TA from limbic temporal lobe seizures are presented in table 2.
Myoclonic seizures (MS)
MS in IGE are sudden, brief, bilateral symmetrical or asymmetrical clonic movements of distal, regional or axial muscles with varying intensity, singularly or organised in rhythmic but more frequently arrhythmic volleys. They mostly occur spontaneously, mainly soon after awakening, or in response to well-known triggers such as flickering lights. They may also occur in association with movements or intention of movements (praxis-induced), or in response to simple or complex (e.g. reading) stimuli, and some of these triggered MS may be unexpectedly local, leading to a misdiagnosis of focal epilepsy (Wolf, 2017; Abarrategui et al., 2018) (table 3). Characteristically, MS occur in clear consciousness and can be remembered and described by the patients. However, a “brain fog” may be described, especially during repetitive clusters of myoclonic seizures, such as in myoclonic-absence status, a rather rare seizure type mostly encountered in JME and MAE patients.
MS frequently occur in some association with TA or GTCS (as in most patients with JME and in some with JAE) or may be the only seizure type, such as in benign myoclonic epilepsy in infancy and in some patients with JME. Bilateral upper limb MS can also occur in some patients with the syndrome of Jeavons (eyelid myoclonia with or without absences), reflecting some overlap between the two syndromes. Asymmetrical or even focal unilateral myoclonic jerks are not uncommon and may typically result in the misdiagnosis of JME as focal motor epilepsy (Usui et al., 2005).
From the EEG viewpoint, MS are characterised by brief generalised polyspike-and-wave discharges with varying intra-discharge frequency and in most cases with an anterior maximum, although higher amplitudes in the occipital region are also observed, especially in patients with eye closure sensitivity or photosensitivity. Discharges may be symmetrical or show variable side emphasis (figure 7). EMG polygraphy is important to show the correlation between the polyspike-wave discharge and the muscle contraction, which sometimes is followed by brief inhibition manifested by fleeting loss of tone. This myoclonic-atonic sequence can only be shown if there is baseline muscle tone, i.e. when the limb is held against gravity (figure 8) and should not be confused with epileptic negative myoclonus that typically occurs in focal structural epilepsies, idiopathic focal epilepsies (where it is typically AED [antiepileptic drug]-induced) and epileptic encephalopathies.
Practical clinical diagnostic clues for generalised myoclonic seizures
MS are the defining seizure type of JME, the most frequent syndrome of IGE, and their clinical suspicion is very important. Differential diagnosis includes focal epileptic myoclonus, i.e. focal motor seizures (a commonly made error, as MS in IGE are often lateralised or asynchronous) and non-epileptic myoclonus of various causes. Clinical clues in favour of idiopathic generalised MS include occurrence on awakening, facilitation by sleep deprivation and identification of possible triggers (lights, reading or praxis). It is important to remember that young patients with JME may not report mild MJ in the belief that it is “normal” to twitch on awakening after a late night out and little sleep. Questions, such as “are you clumsy in the morning, dropping things or spilling your coffee”, helped by physical demonstration and by drawing a parallel with the (physiological) hypnagogic jerks to ensure that patients understand what they are asked may lead to diagnosis (Panayiotopoulos, 2007). As already mentioned, obtaining positive evidence for MS in a patient presenting with a generalised convulsion strongly argues for the latter being GTCS rather than focal to bilateral tonic-clonic, and for the diagnosis of JME.
In summary, there may be significant difficulties and diagnostic pitfalls that make clinical diagnosis at first visit variably uncertain. However, these difficulties should not dishearten the young clinicians in their quest to gather a proper and as complete as possible seizure anamnesis, which will help tailor the diagnostic EEG accordingly, increasing its diagnostic yield (Koutroumanidis et al., 2017).As in other epilepsies, explaining seizure semiology and educating the family on what they should keep observing will improve seizure description in the follow-up visits. Once the diagnosis of IGE is established, it is always useful to ask about prolonged episodes of confusion that may reflect episodes of absence status.
Electroclinical approach and the role of EEG as a diagnostic tool in IGE
Electroclinical correlation is the cornerstone of correct diagnosis and classification of epilepsy and epilepsy syndromes. The EEG can provide crucial information either for a correct syndrome diagnosis or for assessing the clinical relevance (ictal? interictal? subclinical?) of paroxysmal EEG discharges (particularly when EEG is coupled with additional recording parameters such as polygraphic EMG channels). The identification of clusters of EEG features (such as background activity, morphology and topography of interictal and ictal epileptiform discharges, the response to activation procedures and the influence of sleep on epileptiform discharges), associated with a clinically defined epileptic condition, has significantly contributed to the introduction of the “electroclinical concept” of epilepsy syndrome. Indeed, the role of the EEG as an essential tool for the classification of seizures and epilepsies has been recognized in all ILAE classification proposals (Gastaut, 1969; Commission on Classification and Terminology of the ILAE, 1981, 1989), including the recent position papers on classification of the epilepsies by the ILAE Commission for Classification and Terminology (Scheffer et al., 2017) and the recentoperational classification of seizure types (Fisher et al., 2017). It is important to remember that the EEG is not a standard static test, but can be tailored to the diagnostic hypothesis in the individual patient. For instance, IGEs are well known for their sensitivity to standard activation methods, such as hyperventilation, photic stimulation and sleep deprivation, sleep and awakening, while in some, seizures may also be subject to triggers, such as reading, praxis, fixation-off, or thinking, which can be tried during the recording under controlled and safe conditions. It follows that, for highest diagnostic yields, the EEG request should contain not only the provisional clinical impression to be tested, but also circumstances of seizure occurrence and possible triggers when these are suspected (Matsuoka et al., 2000).
Solid understanding of the uses and limitations of the EEG is of paramount importance for clinicians that care for patients with epilepsy, and a full discussion on IGE in particular can be found elsewhere (Koutroumanidis and Smith, 2005).
In summary, the EEG can:
- –Support the diagnosis of IGE and assist in its differential diagnosis involving:
- •symptomatic focal epilepsies with fast generalisation;
- •symptomatic generalised epilepsy.
- –Delineate the full clinical picture, by video recording, of all seizure types and variations, with implications for:
- •a particular IGE syndrome diagnosis;
- •optimal selection of AEDs (i.e. levetiracetam, valproic acid [in males] or clonazepam-when MS are prominent; lamotrigine if TA predominate).
- –Detect specific triggers or self-induction (for instance, in patients with photosensitivity).
- –Diagnose non-epileptic conditions that mimic IGEs (i.e. staring attacks or hyperventilation-induced high-voltage rhythmic slowing mimicking TA, or jerks not associated with GSW discharges).
- –Assist in prognostication (i.e. CAE has better prognosis than other absence syndromes in childhood or JME).
- –Monitor AED treatment and predict possible relapse after AED discontinuation (.mainly in children with TA and patients with photosensitivity [Ambrosetto and Tassinari, 1987])
- –Detect/confirm new seizure types that may signal either evolution of natural history or adverse effects of AEDs.
- –Detect signs of AED intoxication.
- –Record previously unidentified seizures/interictal patterns/triggers when reconsidering initial diagnosis and reclassification after treatment failure.
Interictal EEG (an EEG recording which has not captured a seizure or “event”) alone cannot be used for:
- –Establishing or excluding the diagnosis of epilepsy (including IGEs).
- –Providing oversimplified clues for reliable syndromic diagnosis (GSW do not always indicate IGE, and focal changes do not necessarily suggest a structural focus).
- –Prognostication and prediction of possible relapse after the discontinuation of AED treatment.
EEG features of IGE
The generalized spike-wave discharge (GSWD) is traditionally considered as the EEG hallmark in IGE. However, the term “generalized” does not mean that the discharge starts at the same time across all brain areas. It simply describes the EEG phenomenology, distinctive to IGE, in which the GSWD involves all areas of the cerebrum, either apparently symmetrically with bilateral synchrony, or with variable side or regional emphasis. Polyspikes and polyspike-wave discharges are also not uncommon, especially in non-rapid eye movement (NREM) sleep. Other EEG phenomena, such as generalised photoparoxysmal responses, fixation-off sensitivity, eye-closure sensitivity, epileptic K-complexes and occipital intermittent rhythmic delta activity (OIRDA), may also be seen depending on the EEG recording conditions and the features of the particular syndrome. Incidental focal and/or lateralized features are not unexpected.
Activation methods
Intermittent Photic Stimulation(IPS)is a standard activation method with substantial translation for clinical and research purposes. The technique is relatively well standardized globally (Kasteleijn-Nolst Trenité et al., 2012). Photosensitivity and photoparoxysmal responses (PPRs) occur in several epileptic syndromes, most of which belong to the IGE group with rare exceptions such as focal syndromes, e.g. idiopathic occipital photosensitive epilepsy and other generalized epilepsies such as progressive myoclonic epilepsies. Therefore, PPRs, with the aid of accompanying signs and symptoms, can be supportive evidence for the diagnosis of IGE. PPRs are most commonly encountered in JME, followed by other IGE syndromes (Sadleir et al., 2009). As well as their contribution to the diagnosis, PPRs are also helpful in determining the required lifestyle changes andfollowing up the treatment response.
Eye closure sensitivity is defined as non-coincidental emergence of GSWD within 1-3 seconds of voluntary eye closure that lasts for 1-4 seconds. Eye closure sensitivity commonly accompanies photosensitivity, although it may also be observed independently. It has a particular diagnostic role in EMA but may be also encountered in other IGE syndromes (Fabian and Wolf, 1987; Appleton et al., 1993; Sevgi et al., 2007).
Fixation-off sensitivity (FOS) is the precipitation of interictal and rarely ictal discharges in response to elimination of central vision and fixation. The important feature of FOS, differentiating it from eye closure sensitivity, is the persistence of discharges throughout the whole period with closed eyes and disappearance on eye opening with fixation (Panayiotopoulos, 1998). Although FOS occurs in IGE patients, it is also observed in other epilepsies and even in people without seizures (Koutroumanidis et al., 2009).
Hyperventilation (HV)is extremely successful in activating 3-Hz GSWD and absences. However, some IGE patients may lack this response to hyperventilation (Dalby, 1969; Koutroumanidis et al., 2008).
Sleep and sleep deprivation are very important activation methods in suspected IGE, which is included in the so-called “sleep-related epilepsies”. NREM sleep, and in particular, phase shifts, arousals and drowsiness, has an activating effect on discharges in most IGE patients, whereas REM sleep tends to supress them (Degen et al., 1987; Halász et al., 2002). Awakening, in particular, is a potent activator of GSWD, therefore sleep EEG should also include a period after awakening when HV and IPS may be performed or repeated (Elmali et al., 2017). Epileptic K complexes are mostly seen in patients with IGE (Seneviratne et al., 2016).
Main IGE syndromes
Childhood absence epilepsy (CAE)
Although absences may occur in combination with other seizure types in any genetic generalized epilepsy, they are the most prominent, and in the majority of cases, the only seizure type in patients with absence epilepsies. The onset of absence epilepsy is age-dependent with two peaks, one at age 6-7 and another at around 12 years (Guerrini et al., 2019). Together with clinical differences in clinical presentation and prognosis, this age dependency leads to the differentiation of CAE and JAE.
CAE is one of the most common epilepsy syndromes in children, accounting for 10 to 17% of all epilepsy cases in school-aged children (Berg et al., 1999). Its incidence was found to be 5.8 to 7.1 per 100,000 persons (Olsson, 1988; Sidenvall et al., 1993), leading to a prevalence of 0.4 to 0.7 per 1000 children (Jallon and Latour, 2005).
CAE typically starts between four and 10 years of age with a peak at around six years. At the time of diagnosis of CAE, typical absence is typically the only seizure type. Younger or older age at onset is possible but diagnosis should be made with care with respect to other syndromes. Girls are more frequently affected than boys. The incidence of CAE is 6.3-8/100,000 children aged 0 to 15 years and accounts for 2-12% of all epilepsies (Berg et al., 2000). The typical absences are brief with a sudden onset and loss of awareness, together with a behavioural arrest and fast reorientation. The duration ranges between four and 30 seconds with most absences lasting five to 15 seconds. The median duration is 10 seconds and clinical signs typically start 1.5 seconds after onset (Kessler et al., 2017). Pausing and staring are prominent in 99% of the patients, while motor automatisms occur in 86% and eyes are involved in 76.5%.
In contrast to other IGEs with absences, the seizures in CAE occur at a high frequency, with up to 100 or more absences per day. These can reliably be triggered by hyperventilation in more than 80% of the patients, and photosensitivity can be expected during EEG recording in up to 20% of the patients (Sadleir et al., 2009). Bilateral symmetric spike-wave discharges start with a frequency of 3-3.5 per second and typically slow down to 2.5-3/second during the seizure. Within the seizure pattern, polyspikes or variations in frequency can sometimes be observed. The amplitude has a frontocentral maximum but occipital predominance is also possible.EEG background activity is normal in most and only mildly slower in some cases, and occipital intermittent rhythmic delta activity can be seen in about 25% (Seneviratne et al., 2017). A coincidence of CAE with the genetic marker of centrotemporal spikes (Hedström and Olsson, 1991) and even the combination with rolandic epilepsy have been described (Verrotti et al., 2017). Spike-wave discharges appear more fragmented and slower during sleep and may be accompanied by polyspikes (Sadleir et al., 2009).
A minority of patients suffer from GTCS that typically start years after CAE onset (Callenbach et al., 2009), although others found higher rates up to 69% with occurrence of GTCS, five to 10 years after the onset of absences, and their co-occurrence had a negative prognostic significance (Grosso et al., 2005). Myoclonic seizures are rare and should lead to a diagnostic revaluation.
Patients with CAE develop normally without any neurological abnormalities. However, subtle cognitive deficits can be seen in 25% of patients and psychiatric comorbidities (ADHD, anxiety disorder and others) are present in about 60% (Caplan et al., 2008). Based on a study by Caplan et al., risk factors were reported to be longer epilepsy duration, higher seizure frequency and treatment with AEDs (Caplan et al., 2008). Even immediately after diagnosis, Shinnar et al. detected elevated total CBCL scores in about 8% of children with CAE (Shinnar et al., 2017). In the same cohort, Connor's Continuous Performing Test was pathologic in one third of the newly diagnosed and untreated children with otherwise intact cognitive functioning (Masur et al., 2013). These difficulties persisted after 16 to 20 weeks even in patients who became seizure-free. This should lead to screening for attention deficit in all patients. In addition, patients with CAE are at risk of life-long psychosocial adaptive problems (Wirrell et al., 1997).
The long-term seizure prognosis of CAE is very good. In patients rendering seizure-free within the first months after diagnosis, long-term remission can be expected in more than 80%, while the remission rate in patients with an initial refractory course and seizure freedom achieved after six months is below 40% (Callenbach et al., 2009). Remission before adulthood can be expected in up to 90% of patients with absences only (Guiwer et al., 2003).A worse prognosis is associated with an atypically early (10 years) onset, early refractory course and photosensitivity (Guerrini, 2006). Further factors associated with worse outcome were absence status, older age at onset (>8 years), focal EEG abnormalities including epileptic discharges, and abnormal background EEG (Wirrell et al., 1996; Grosso et al., 2005). Evolution to JME was reported in 5 to 15% of patients with CAE (Wirrell et al., 1996; Trinka et al., 2004). Despite the good long-term prognosis of CAE in the majority, psychosocial adaptation is poor in many patients (Wirrell et al., 1997).
Juvenile absence epilepsy (JAE)
In JAE, seizure onset occurs around puberty and rarely in younger patients. In contrast to CAE, frequency of absences is much lower with several seizures a day or even less and ictal loss of awareness may be less severe. Absence status can occur. Up to 80% of patients suffer from GTCS which can be the initial symptom. Spike-wave discharges have a frequency of four to five per second at absence onset. Irregular and disorganized spike-wave discharges are eight times more likely than in CAE (Sadleir et al., 2009). Provocation of absences by hyperventilation is as frequent as in CAE (>80%) and about 25% of the patients are photosensitive (Sadleir et al., 2009). EEG background activity is normal in most cases. All patients present with fragmented spike-wave discharges and polyspikes that occur mostly in drowsiness and/or sleep (Sadleir et al., 2009). Sleep deprivation is a prominent trigger of absences and other seizure types in JAE.
Response to antiepileptic treatment with complete seizure control can be expected in only 15% (Danhofer et al., 2014) to 60%(Trinka et al., 2004) of the patients and long-term prognosis is poor regarding complete remission (Wirrell et al., 1996). Most patients require life-long antiepileptic therapy. Absences may occur less frequently over time but GTCS persist in many patients.
Differential diagnosis of CAE and JAE
The major differences between CAE and JAE are outlined in table 4. Absences starting at atypical age should lead to careful differential diagnostic considerations. Early-onset absence epilepsy, starting before four years, is caused by GLUT1 deficiency in 12% of the cases (Suls et al., 2009). This aetiology should be ruled out by lumbar puncture and/or analysis of the SLC2A1 gene. The diagnosis requires a personalized therapy with a ketogenic diet.
Other syndromes, for example “eyelid myoclonia with absences” (Jeavons syndome), have been described but were not considered by the ILAE in the latest classification.
Juvenile myoclonic epilepsy (JME)
JME is one of the most common genetic/idiopathic generalized epilepsies. Onset is between eight and 25 years of age. Both sexes are equally affected. A small number of JME cases (around 5%) evolve from CAE. Developmental milestones, cognition and neurological examination are normal. Mandatory seizure types for the diagnosis are myoclonic seizures, prominent at the upper limbs (Oguni et al., 1994), which are seen especially on awakening (30 minutes to one hour from awakening) (Touchon, 1982). Other seizures that can be observed in JME are generalized tonic-clonic seizures (in more than 90% of subjects), often heralded by a build-up of myoclonic jerks, and absence seizures (in about 30% of individuals), usually less frequent, shorter (on average 3 seconds), and with less impairment of consciousness than in CAE (Genton et al., 2013; Kasteleijn- Nolst Trenité et al., 2013).
Interictal EEG shows a normal background activity and may display generalized spike-and-wave and polyspike-and-wave (usually at 3.5-6 Hz) discharges. Fragmented generalized spike-and-wave bursts are not uncommon and can appear as focal or multi-focal, but usually not consistently seen in a single area. Focal spikes consistently seen in a single area should raise the suspicion of structural brain abnormality, whereas a slow-spike-and-wave (Serafini et al., 2013; Koutroumanidis et al., 2017).
Intermittent photic stimulation can elicit a photoparoxysmal response in one third of cases, although visually-induced seizures in the daily environment are reported more rarely (in about 10% of individuals) (Janz, 1985; Wolf and Goosses, 1986). Hyperventilation can induce generalized spike-and-wave or polyspike-and-wave patterns and even clinical absences. Sleep deprivation can enhance EEG abnormalities, especially PPRs.
Ictal EEG is characterized by a generalized polyspike-and-wave pattern, associated with myoclonic seizures; a single generalized polyspike-and-wave complex can elicit a myoclonic jerk. Absence seizures are associated with regular, generalized spike-and-wave or polyspike-and-wave discharges, usually faster (3.5-6 Hz) than in CAE. During generalized tonic-clonic seizures, fast rhythmic spikes can be seen in the tonic stage; bursts of spikes and after-coming slow waves are synchronous with clonic jerks (Serafini et al., 2013; Koutroumanidis et al., 2017).
To be considered in the differential diagnosis:
- –JAE: evidence of prominent myoclonic seizures distinguishes JME from JAE.
- –Epilepsy with eyelid myoclonia. This syndrome should be suspected in case of repetitive, very frequent, episodes with fast (>4-Hz) rhythmic jerks of the eyelids and upward deviation of the eyeballs.
- –Epilepsy with myoclonic absences: its distinctive features are myoclonic jerks in the upper limb at 3 Hz associated with a concomitant tonic contraction raising the arms; seizures are usually longer than MS in JME.
- –A structural brain abnormality when myoclonic or generalized tonic-clonic seizures and EEG show consistent focal features among seizures.
- –Progressive myoclonus epilepsies: this heterogeneous group of diseases is characterized at variance with JME by intractable myoclonus, cognitive decline, neurological impairment, and slowing of the background on EEG.
JME is considered a life-long condition and most clinicians are reluctant to withdraw antiepileptic treatment. However, more recent literature suggests a heterogeneity in the disease course, and life-long treatment might not be a necessity for all patients (Geithner et al., 2012; Kasteleijn- Nolst Trenité et al., 2013). In the study of Camfield and Camfield (2009), (17)% of patients, a small but significant number, were able to discontinue medication and remain seizure-free 25 years after seizure onset. Another 13% experienced only myoclonic seizures, therefore, AED treatment was no longer used by one third of patients in this JME population. Other studies have also shown that although most JME patients had continuing seizures in the long term, the dominating myoclonic seizures diminish or are at least distinctly alleviated in the fourth decade of life, and the disease tends to stay under control with an appropriate antiepileptic regimen (Baykan et al., 2013).
IGE with GTCS alone (GTCS-a)
As the name indicates, GTCS are the defining seizure type; typical absences and myoclonic seizures (either as part of the clinical picture or identified on EEG) are not constituents of this syndrome. Therefore, diagnosis of GTCS-a in patients with history of GTCS and GSWD on EEG effectively relies on the exclusion of both myoclonic seizures and typical absences (including phantom absences), and by implication, on the extent and thoroughness of the initial clinical and EEG assessment and subsequent clinical-EEG follow-up (Koutroumanidis et al., 2008). On the other hand, diagnosis of IGE with GTCS alone cannot be made in the absence of GSWD on the EEG (Scheffer et al., 2017). Some patients with focal epilepsy, who report only “GTCS”, pose a significant challenge in the differential diagnosis; therefore, patient history, seizure semiology and EEG recordings should be scrutinized for any possible atypical or focal findings.
The age at onset of GTCS-a ranges from childhood to mid adulthood, peaking during the second decade. GTCS typically occur within 1-2 hours of awakening but may also infrequently occur during periods of relaxation and in some patients randomly. Sleep deprivation, early forced awakening particularly after short or poor sleep, tiredness and stress are principle precipitants (Janz, 2000; Unterberger et al., 2001). The syndrome is not self-limiting; response to antiepileptic medication is in most patients satisfactory, although efficacy is difficult to ascertain in patients with infrequent GTCS. Indeed, seizure frequency is generally low (Camfield and Camfield, 2010; Holtkamp et al., 2014).
Atypical clinical presentations
On clinical neurological examination, few abnormalities are expected in IGE patients and if any are encountered, they are either coincidental or point to a more complex syndrome. Absences, myoclonic seizures, GTCS or any combination of these seizures in an otherwise healthy child, teenager or young adult should suggest IGE as an initial diagnosis. However, some cognitive or behavioural features may be striking.Some children with mild absences may be diagnosed with learning difficulties or mild intellectual disability.These cognitive difficulties are a mere consequence of lapses of attention during the seizures rather than a true cognitive dysfunction in most of the cases. A very early onset of absence seizures can be the hallmark of GLUT1-deficiencey which seems to be the aetiology in about 10% of all cases with early-onset absence epilepsy (Suls et al., 2009). The combination of microcephaly, developmental delay, ataxia and generalized seizures should, on the one hand, lead to the exclusion of GLUT1-deficiency and, on the other hand, remind us of various PME including neuronal ceroid lipofuscinosis and mitochondrial disorders. In older children and adolescents, a combination of new-onset generalized seizures (i.e. MS) with neurological (vertical gaze paresis, ataxia and others) and/or psychiatric symptoms is suggestive of either autoimmune encephalopathy or PME including various storage diseases (Niemann-Pick type C and others).
Behavioural problems and personality traits havent been associated with IGE syndromes. Executive dysfunction, especially impulsiveness, is a very well-known feature accompanyingJME. Other psychosocial complications, such as ADHD or impaired facial emotional recognition, may also accompany the seizures, raising the question whether the spectrum for IGE goes beyond just a seizure disorder (Gomez-Ibañez et al., 2014; Almane et al., 2019; Syvertsen et al., 2019). A comprehensive neuropsychological evaluation of these patients may enhance our knowledge about GGE/IGE and enable each individual to reach their full potential.
Imaging for IGE
The evidence for the value of systematic usage of structural MRI in GGE/IGE groups is currently insufficient. The most important utilization of structural MRI is to differentiate between focal epilepsies mimicking generalized syndromes and true GGE/IGE cases. The current recommendation is using HARNESS-MRI protocol (the Harmonized Neuroimaging of Epilepsy Structural Sequences) if the patient presents with atypical features, namely developmental and cognitive issues, persistent seizures, focal EEG findings and abnormal neurological examination findings (Bernasconi et al., 2019). It has to be emphasised that incidental structural changes should not distract physicians from the diagnosis of IGE in patients with otherwise typical electroclinical presentation.
Genetics
Although recently named “genetic”, generalized epilepsies usually lack clear mutations as in conventional genetic diseases. Contribution of genetic variants is not straightforward, and the clinical picture emerges from the interplay between genotype and environmental factors, a fact suggested by Lennox as early as the 1940s based on his valuable twin studies (Lennox, 1947). For GGE/IGE, the concordance rate in monozygotic twins is 76% and for dizygotic twins 33% (Berkovic et al., 1998). Even in the light of recent advances in genetics, only a handful of genes with monogenic inheritance have been associated with IGE, most of which encode for vital parts of channels in the central nervous system. There are also variants associated with an increase in susceptibility to IGE, however, the current knowledge suggests that multiple variants are necessary for an IGE phenotype. A model explaining the polygenic nature and interaction of these variants is currently lacking (Mullen et al., 2018). In the light of current knowledge, genetic testing has very few clinical applications in routine clinical practice for IGE patients and should not be utilized on a routine basis. Readers can find more information in the supplementary material.
Conclusion
In conclusion, idiopathic generalized epilepsy is a colourful spectrum comprising a number of distinct syndromes. However, in real-life situations, clinical history and EEG findings can sometimes intertwine and make it hard, if not impossible, to classify individual patients within a specific syndrome. A multidimensional approach and keeping an open mind as new information becomes available are key factors in diagnosing and treating these patients appropriately.
Key points
- •Correct diagnosis and classification of IGE syndromes is based on detailed history, physical examination and congruent EEG findings.
- –Age of onset, seizure type, frequency, timing and triggers should be questioned.
- –No neurological signs or neuroimaging findings are expected and existence of such must be evaluated as red flag findings.
- •GTCS have a sudden onset, loss of consciousness is instant, last 40-70 seconds and recovery is gradual.
- •Typical absences last 2 to > 20 seconds and are characterized by a sudden impairment of consciousness with an abrupt ending. They are effectively triggered by hyperventilation.
- •Myoclonic seizures are sudden, brief, symmetrical or asymmetrical clonic movements generally occurring with clear consciousness.
- •Incidental neuroimaging findings should not distract the physicans from IGE diagnosis in cases with otherwise typical electroclinical presentation.
- •Genetic testing has very little clinical significance for most IGE patients and therefore should not be utilized on a routine basis.
Case 1
A 21-year-old woman was evaluated following a GTCS. This GTCS occurred shortly after morning awakening, following a late night out with friends and little ensuing sleep. Her flat mate witnessed the event and described it as follows:
“She was drinking her coffee and I was preparing the breakfast. I heard a scream and when I looked at her, I saw her eyes were wide open. Her arms have risen up, she became stiff and after about 10-20 seconds, she started shaking violently. This lasted about a minute, she wetted herself in the end and afterwards she slept for an hour before gradually coming round.”
Upon further questioning, the patient said that she would have jerks prominently in her arms and upper body when exposed to flickering lights and she would be quite clumsy in the morning (she had been dropping things from her hands over a couple of years), but she just assumed that this is the same for everybody. She admitted she was more prone to take risks and easily irritable compared to her peers. She recalled being told that her grandmother had seizures as a teenager and young adult but she did not know more details.
Her sleep-deprived EEG revealed 3.5-6-Hz generalized spike wave and polyspike wave discharges, some of which were accompanied by myoclonic jerks. She was diagnosed with JME.
Case notes:
A JME case is described. Typical triggers (sleep deprivation, photosensitivity, awakening) are quoted by the patient in the history. Morning clumsiness due to jerks have been overlooked by the patient for years assuming they were normal, as happens in most cases. A GTCS is described beautifully by the flat mate. Typical personality traits associated with JME are also noted. There is a positive family history in keeping with the genetic background of the disease, and the EEG revealed typical findings.
Case 2
A 15-year-old boy had been diagnosed with focal epilepsy and learning difficulties and treated with carbamazepine. His mother says that his seizures started at around the age of 10, and describes that during these, he abruptly stops whatever he is doing and displays some aimless movements such as rubbing his nose or smacking lips, for a while; he then continues as if nothing has happened. There have been a few times when he looked vacant and was not quite himself for a couple of hours; some of these long states ended with “big seizures”, during which he stiffens and jerks. His learning difficulties started with his seizures and the patient states that he misses chunks of the school lectures and it is difficult for him to follow the class.
The mother herself suffered from vacant spells as a little girl and had to take valproic acid for a couple of years but she has been symptom-free for a long time.
The boy's EEG revealed 3-Hz spike-wave discharges. During hyperventilation with breath counting, he was noted to pause during discharges and continue immediately after. Longer seizures were associated with a potpourri of automatisms. Diagnosis changed to juvenile absence epilepsy and a switch to valproic acid has led to complete resolution of both his seizures and his “learning difficulties”.
Case notes:
This is an example of juvenile absence epilepsy with some pitfalls. Accompanying automatisms lead to a misdiagnosis of focal epilepsy and prescription of carbamazepine which in turn possibly aggravated the absence seizures. The long vacant periods are suggestive of absence statuses. Learning difficulties, which would suggest an other aetiology, are most likely due to lapses of attention during frequent absences.
Age at onset, abrupt onset and cessation of seizures, and the mother's history suggestive of childhood absence epilepsy are the key points leading the clinician to the correct diagnosis, confirmed by the typical EEG findings.
Supplementary data
Summary didactic slides and supplementary material are available on the www.epilepticdisorders.com website.
Disclosures
None of the authors have any conflict of interest to declare.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International