Epileptic Disorders
MENUEpilepsy of infancy with migrating focal seizures: three patients treated with the ketogenic diet Volume 17, numéro 2, June 2015

Epilepsy of infancy with migrating focal seizures (EIMFS) is a well-defined and rare epileptic syndrome, characterized by an onset of multifocal seizures before 6 months of age and a typical ictal EEG pattern, consisting of seizures that arise independently and sequentially from both hemispheres (Coppola et al., 1995). The seizures are refractory to antiepileptic drugs (AEDs) and cause subsequent severe intellectual disability (Coppola et al., 1995; Caraballo et al., 2008).
Several reports have focused on seizure control or reduction with the use of different AEDs, such as levetiracetam, adrenocorticotropic hormone (ACTH), benzodiazepines, stiripentol, rufinamide, and potassium bromide, alone or in combination, with variable results (Caraballo et al., 2008; 2014a; Djuric et al., 2011; Vendrame et al., 2011).
Recently, EIMFS has been described to be related to different genes in sporadic and familial cases (Lee et al., 2012; Poduri et al., 2013; Striano et al., 2014), suggesting genetic heterogeneity.
The ketogenic diet (KD) has been successfully used in children with a variety of seizure types and epileptic syndromes, especially epileptic encephalopathies (Kossoff et al., 2008; Caraballo et al., 2011). Here, we present three patients with EIMFS and provide a comprehensive evaluation of KD therapy.
Case studies
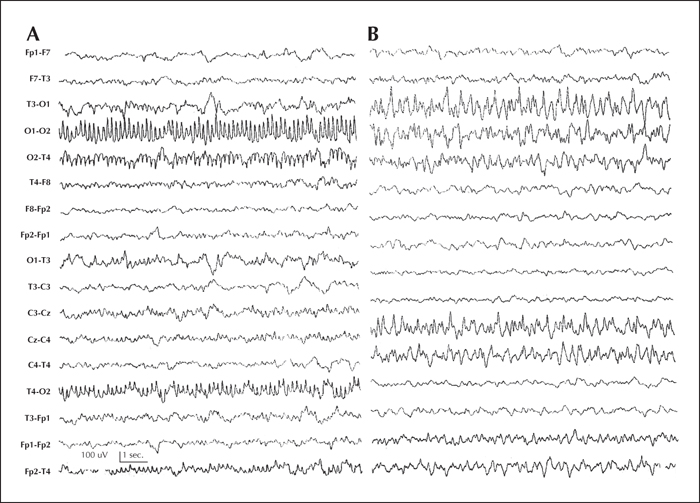
Patient 1 was a 20-month-old boy with migrating focal clonic seizures from 20 days of life, either alternating between hemispheres or affecting a single hemisphere, characterized by clonic facial movements, respiratory changes, and cyanosis. The first interictal EEGs did not show significant alterations. During follow-up, the seizure frequency increased, with a migrating pattern (figure 1 A and B) and status epilepticus. MRI, metabolic screening, and karyotype were normal.
The boy received phenobarbital, carbamazepine, lorazepam, valproic acid, and levetiracetam in different schemes, with partial response. At eight months, he developed epileptic spasms. Vigabatrin was added initially with a good response. However, a month later, he had brief focal seizures characterized by eye deviation and desaturation. He developed hypotonia, microcephalus, and visual disturbances. At 13 months of age, the KD was started and the patient remained seizure-free after seven months of follow-up, with excellent tolerance of the diet. Two of four AEDs could be discontinued. The video-EEG recording did not show any ictal events. Neurocognitive development significantly improved.
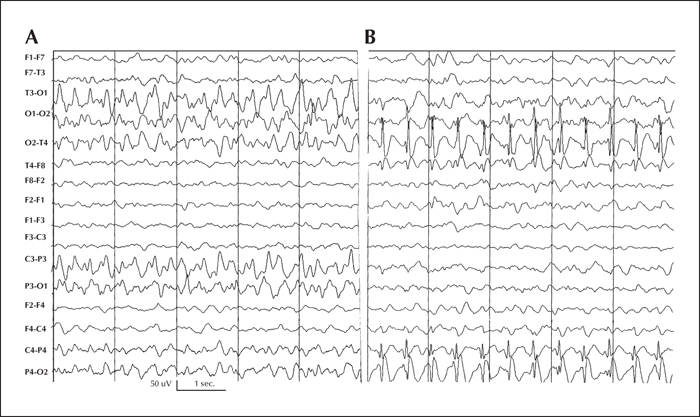
Patient 2 was a 29-month-old boy with weekly focal clonic seizures from 30 days of life. At two months of life, the focal seizures significantly increased and additionally he developed complex focal migrating seizures, either alternating between hemispheres or affecting a single hemisphere. The first interictal EEGs showed a mildly disorganized background rhythm without paroxysms. The seizures, showing the same migrating pattern and frequent status epilepticus, became refractory to AEDs, requiring prolonged hospitalisation and sometimes admission to the intensive care unit. The seizures did not respond to classic and new AEDs, including bromides. An ictal EEG confirmed the characteristic migrating pattern (figure 2 A and B). Repeated brain MRI did not show abnormalities. The boy developed marked hypotonia, microcephaly, and global psychomotor impairment. Metabolic screening and karyotype were normal. No genetic studies for the EIMFS were performed.
At 19 months of age, the KD was started with a partial response. Seizure frequency decreased by less than 50%, but mild improvement of neurocognitive development was observed and his quality of life improved after 18 months on the KD, associated with levetiracetam and topiramate treatment. During the period on KD, the AEDs were reduced from four to two. The last video-EEG recording showed a significant reduction of the ictal events.
Patient 3 was a 4 year, 10-month-old girl who had isolated seizures during sleep at three weeks of life. The episodes repeated one month later. A month and a half later, seizure frequency markedly increased to 200 episodes, daily. The seizures were characterized by eye and head deviation and eyelid twitching, followed by eye and head deviation to the opposite side and chewing movements. Different AEDs, such as topiramate, clobazam, vigabatrin, phenytoin, and phenobarbital, were tried, but the girl did not respond to any.
The EEG showed unilateral slow activity that migrated to the other hemisphere and disappeared in the initially affected hemisphere. MRI, CT, and neurometabolic screening were normal. Two months later, the introduction of levetiracetam reduced seizure frequency from 25 to 30 daily episodes during sleep. Seizures further reduced to 20, a day after adding zonisamide, one year and three months later. The patient developed microcephaly, hypotonia, language impairment, and eye-tracking difficulties. MRI showed cortical and central atrophy. Three years after seizure onset, while on valproic acid, levetiracetan, zonisamide, and lorazepam, seizure frequency again increased, and at 19 months of age, the KD was started. Seizures reduced from five to seven a day and some days she was seizure-free. Currently, the girl continues on the diet after 20 months with a seizure reduction of 75-99% and excellent tolerance of the diet. While on the KD, the number of AEDs was reduced to two. The video-EEG recordings, as well as neurocognitive development, have significantly improved.
Discussion
We describe three patients who presented with AED-refractory EIMFS before 5 months of age and who were treated with the KD. Two of them responded very well to the diet; one became seizure-free and the other had a seizure reduction of 75-99% after 8 and 18 months of follow-up, respectively. In these two children, neurocognitive development significantly improved. The third patient had a seizure reduction of less than 50%, but in this patient, neurocognitive performance moderately improved. In all three patients, the KD was well tolerated.
In EIMFS, seizures are markedly drug-resistant and outcome is generally poor. Nevertheless, some patients may respond favourably to bromides, ACTH, stiripentol associated with clonazepam, and levetiracetam (Caraballo et al., 2008, 2014a; Djuric et al., 2011; Vendrame et al., 2011). In addition, vagus nerve stimulation and the KD have also been tried, but with, up to now, poor or inconclusive results (Coppola, 2013).
The KD has been successfully used for children with a variety of encephalopathies, including Lennox-Gastaut syndrome, West syndrome, epilepsy with myoclonic atonic seizures, Dravet syndrome, Landau-Kleffner syndrome, Tassinari syndrome, and FIRES (Kossoff et al., 2008; Caraballo et al., 2011). EIMFS is included in the group of epileptic encephalopathies that respond well to the KD, as has been demonstrated in two of three cases that were similar to Dravet syndrome, one of the most severe epileptic encephalopathies (Caraballo, 2011). Focal status epilepticus frequently occurs in patients with EIMFS, and recently an excellent response of patients with focal status epilepticus to the KD has also been published (Shein et al., 2012; Caraballo et al., 2014b).
Larger series of patients with EIMFS should be studied with longer follow-up to demonstrate the role of KD in the treatment of this severe epileptic encephalopathy.
Conclusion
The KD is a good option for patients with refractory seizures and should therefore be considered earlier in the treatment scheme for patients with EIMFS.
A better recognition of EIMFS will enable adequate and vigorous treatment to be initiated in order to improve prognosis.
Supplementary Data.
Summary didactic slides are available on the www.epilepticdisorders.com website.
Disclosures
We disclose we have no financial and personal relationships with other people or organizations that could inappropriately influence this work.