Epileptic Disorders
MENUEpilepsia partialis continua associated with the p.Arg403Cys variant of the DNM1L gene: an unusual clinical progression with two episodes of super-refractory status epilepticus with a 13-year remission interval Volume 24, numéro 1, February 2022


- Mots-clés : super-refractory status epilepticus, childhood-onset DNM1L encephalopathy, myoclonus, epilepsia partialis continua
- DOI : 10.1684/epd.2021.1375
- Page(s) : 176-82
- Année de parution : 2022
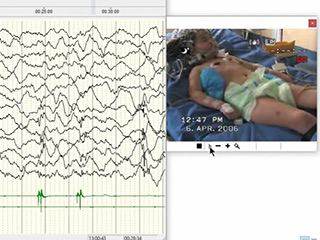
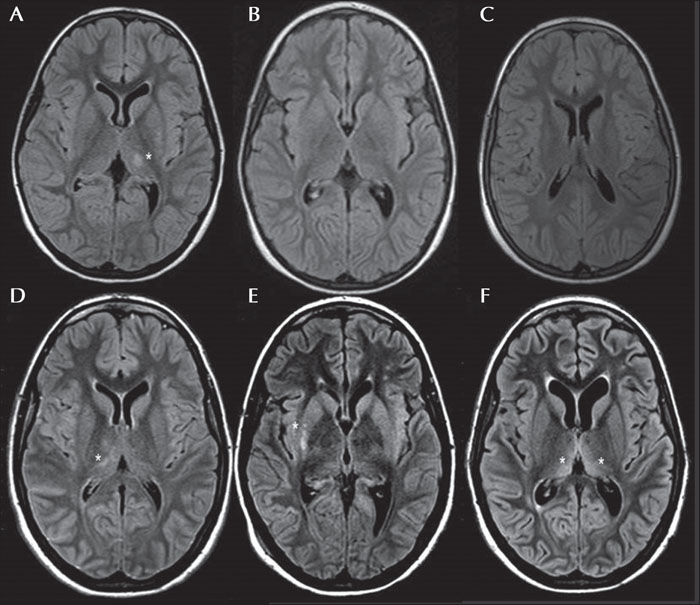
Dynamin-1-like (DNM1L) is a gene located on chromosome 12p11.21 that encodes for dynamin-related protein (DRP1), a GTPase involved in mitochondrial and peroxisomal fusion, which plays a pivotal role in brain development. The missense variant, p.Arg403Cys, is clinically associated with childhood-onset super-refractory status epilepticus, with either subsequent poor neurological outcome or death (described in 13 patients). We present a 20-year-old girl carrying this mutation with a history of two episodes of super-refractory focal myoclonic status epilepticus which manifested as epilepsia partialis continua (EPC) with a 13-year interval, during which she displayed moderate intellectual disability, social and school reintegration, without complete control of myoclonic manifestations. The first status, which occurred at the age of six, was associated with transient left side thalamic involvement and the second episode with right side transient basal ganglia hyperintensity on MRI. After the second status, a persistent vegetative state with both drug-resistant epilepsia partialis continua and reticular myoclonus endured; the MRI showed progressive brain atrophy. In contrast to previous published cases, this new case of childhood-onset DNM1L encephalopathy demonstrated biphasic clinical progression. The main features of our patient were EPC, super-refractory status epilepticus, and transient and migrating subcortical thalamic hyperintensity on MRI at onset. The unusual clinical course is also noticeable, indicating possible epigenetic and/or protective factors, without underestimating the progressive and genetic basis of this encephalopathy. Precise characterization of seizures and whole-exome sequencing are crucial in order to establish early diagnosis.