Epileptic Disorders
MENUCharacteristic phasic evolution of convulsive seizure in PCDH19-related epilepsy Volume 18, numéro 1, March 2016

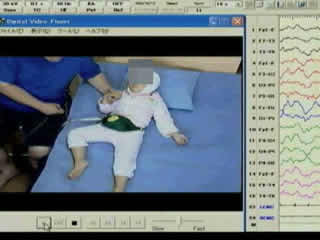
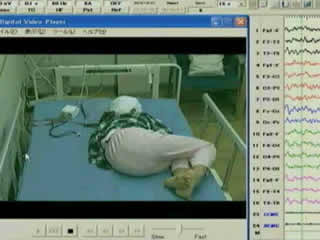

The disorder “epilepsy and mental retardation limited to females” (EFMR) presents seizures in infancy, often in combination with intellectual disability or autism (Juberg and Hellman, 1971; Ryan et al., 1997; Scheffer et al., 2008). Recently, it has been reported that mutations in the X chromosome-encoded PCDH19 gene cause EFMR (Dibbens et al., 2008). As such, the term “EFMR” has been replaced by PCDH19-related epilepsy (PCDH19RE) in recent reports. Subsequent studies revealed sporadic cases with de novo mutations (Jamal et al., 2010; Higurashi et al., 2011; Hynes et al., 2010; Specchio et al., 2011), but the genetic mechanism underlying the phenotypic expression of EFMR remains elusive. The onset of seizure is early and seizures tend to occur in clusters, which are often induced by fever (Higurashi et al., 2011; Specchio et al., 2011; Marini et al., 2012; Higurashi et al., 2013). Here, we report the video-EEG recordings of convulsive seizures in three patients with PCDH19RE, aiming to elucidate the characteristic features of convulsive seizures associated with PCDH19RE.
Methods
Analysis of convulsive seizure semiology was conducted by recording and reviewing ictal video-EEG. Family history, precipitation by fever, frequency and duration of seizures, interictal EEG, brain imaging, treatments, and cognitive and behavioural assessments were reviewed from the medical records. Epileptic seizures were classified according to the International League Against Epilepsy criteria. Genetic analyses of PCDH19 and SCN1A were performed at Fukuoka University, as described previously (Higurashi et al., 2011). The GenBank accession numbers used as reference sequences are the complete human PCDH19 mRNA and protein EF676096.1, and the complete human SCN1A coding sequence and protein AB093548.
The patients and their parents were informed and they consented to participate in this study. The PCDH19 and SCN1A genetic tests were approved by the institutional ethical committee.
Results
Five patients with PCDH19 mutations were identified in our hospital. Among these five patients, we detected 47 seizures by EEG, of which 43 occurred during sleep. Twenty-six convulsive seizures from three patients were recorded by video-EEG and analysed in detail.
Patient 1
Patient 1 was a 14-year-old girl whose mother had severe toxaemia during pregnancy. The patient had no medical history of neurological diseases. The psychomotor development was normal before seizure onset. Her initial seizure occurred at 10 months of age, induced by high fever. Since then, intellectual disability has been observed. Her IQ was 62 at 11 years of age. MRI and computed tomography (CT) revealed slightly diffuse brain atrophy, however, not pathological for her age.
The patient's mother also had epilepsy and intellectual disability. She used to have clusters of seizures similar to her daughter until the age of 31 years when valproate (VPA) was introduced, which she took throughout her pregnancy. VPA was successfully discontinued at the age of 45 years. The patient's father committed suicide due to depression. A PCDH19 missense mutation(c.416C>T/p.Ser139Leu) was detected in this patient and also in her mother. No SCN1A mutation was detected.
The convulsive seizures occurred in clusters with fever. Each seizure lasted for one minute. The seizures recurred every one or two hours, five to 10 times a day. She never had seizures without fever. The patient was treated initially with phenobarbital (PB) at 10 months of age and then carbamazepine (CBZ), but the seizures could not be controlled. When she first visited our hospital at the age of 10 years, we discontinued CBZ and introduced VPA (with concomitant PB). The patient has been seizure-free for two years since then, even when she had a high fever. Interictal EEG showed normal alpha rhythm without paroxysmal epileptic discharges, but with some frontal dominant slow waves. She showed a photoparoxysmal response without clinical correlates by intermittent photic stimulation.
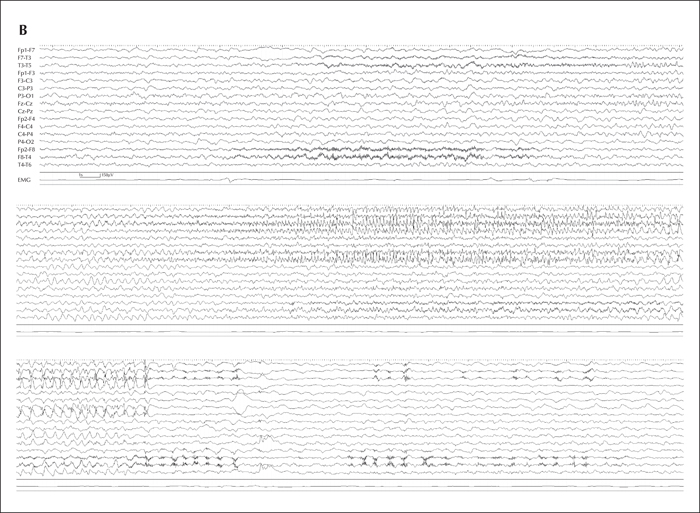
Six convulsive seizures were recorded by video-EEG recording, all of which occurred during sleep. They began with several jerks of the body, resulting in cough-like movement, after which her body and limbs became mildly tonic. The patient's right lower extremity became tonic earlier than her left extremities. She then turned over and both lower extremities showed tonic extension. The right hand and fingers were widely extended, whereas the left hand was flexed. The fingers on both hands and the lower extremities repeatedly moved in a less synchronous, less rhythmic, and less intense manner. The mild trembling/fluttering feature of the seizures was more marked in the distal part of the extremities. Thereafter, this movement gradually became more synchronized, nearly clonic, but less intense, synchronous, and asymmetric. In the postictal state, the patient gasped with oral automatism. Seizures were 1.5 to 3 minutes in duration and tended to repeat every seven to 10 minutes for one to two hours. On ictal EEG (figure 1A), alpha-band waves with a slight predominance of the Cz and C3 regions were followed by rhythmic waves appearing from the Cz, which soon generalised. Another ictal EEG (figure 1B) during the same cluster showed different sequential rhythmic waves predominantly on the left side. Though ictal EEG mimicked typical secondary generalised tonic-clonic seizures (sGTC), surface electromyograms (EMGs) of both deltoid muscles showed only subtle discharges, suggesting that proximal muscles were less involved than peripheral ones (video sequence 1).
Patient 2
Patient 2 was a six-year-old girl who was born after an uneventful pregnancy and had no specific medical history. Her development was normal at 6 months of age, but became retarded after the onset of epilepsy. Her full-scale IQ score was 49 at 6 years of age, and autistic symptoms had also been noted. MRI/CT showed slightly diffuse brain atrophy. This proband's brother, brother-in-law on the mother's side, and cousin on the father's side each had a history of a single febrile seizure. The patient and her father had a PCDH19 missensemutation (c.1787A>T/p.Asp596Val), but this was asymptomatic in the father. Since 6 months of age, the patient had experienced clusters of seizures. The seizures occurred under afebrile conditions initially, but later also under febrile conditions, in a cluster consisting of more than 10 seizures. The seizure clusters occurred a few times a day, and recurred at intervals of three weeks to four months. Despite initial treatment with CBZ and clonazepam (CZP), the seizure cluster repeated. Treatment with zonisamide, clobazam (CLB), and VPA aggravated seizures. Midazolam was given intravenously and frequently until CBZ and CLB were given in combination.
The interictal EEG showed slightly irregular theta rhythms over both occipital-parietal central regions with rare frontal dominant spike waves. Most seizures occurred during sleep and two clusters with six convulsive seizures were recorded by video-EEG. In the first seizure (video sequence 2), the patient was awakened by massive jerks as if startled and started to sit up. Then, she slowly raised both arms tonically while vocalizing. Her eyes deviated first to the right, then to the left, and progressively to the right again. Her pelvis also moved to the right and left. Her right hand was open wide, and her left hand closed. Her limbs and fingers were fluttering or jerking less synchronously, similar to Patient 1. At the end of the seizure, more synchronized jerks were repeatedly observed with a slow rolling movement of the arms. In the postictal phase, the patient mumbled. The seizures usually lasted one minute and tended to occur every 0.5 to 1 hour in a cluster for 12 to 36 hours, usually with high fever. The ictal EEG (figure 2A) started with left posterior temporal sharp transients subclinically. The recruiting rhythms appeared predominantly in the left fronto-temporal region, and then spread bilaterally. In the postictal period, a motion artefact due to postictal automatism was observed on the bilateral temporal leads. The bilateral deltoid EMG did not show marked discharges, but the EEG resembled the sGTC pattern. Another seizure was recorded one year later during sleep. Massive axial jerks occurred first, and then she seemed upset and tried to sit up while vocalizing. Her eyes deviated to the right, and then her face and body turned to the left. All the limbs were extended in a mild tonic manner. The fluttering movements of the hands and fingers were less marked than in the first recorded seizure. After the seizure, she soon fell asleep without postictal automatism. The ictal EEG (figure 2B) began with fast waves emerging predominantly over the right hemisphere.
Patient 3
Patient 3 was 3 years old at the time of the study. She had no remarkable perinatal problems or specific medical history. She had normal development before the onset of the seizures but gradually showed mild intellectual disability and delayed motor development. Her IQ was 49 at the age of 2 years, and she was found to have autism.
The patient had a PCDH19 hemizygous whole-gene deletion, and her aunt on the mother's side had a single febrile seizure. She experienced her first cluster of seizures induced by high fever at nine months of age. From the age of 1 year, she began to have seizures approximately four or five times a day over a two-day period, with each seizure lasting for less than one minute. These clusters recurred four or five times per year with high fever. She was given levetiracetam (LEV), but the seizures repeated. LEV, VPA, oxcarbazepine, and clorazepate were all ineffective. CZP was temporarily effective, but discontinued because of hypersalivation and irritability.
The interictal EEG showed irregular slow waves and independent sharp waves at the left or right frontal region. All 14 convulsive seizures were recorded during sleep, starting with multiple jerks. It appeared as though she was trying to get up but failing to do so, apparently owing to tonic contraction. Her right and left legs were extended sequentially. Both arms were extended tonically, and her eyes deviated to the left. Thereafter, all her limbs and fingers started to jerk rhythmically and synchronously in a mild form, similar to Patients 1 and 2. In the postictal phase, oral automatism was observed. Each seizure lasted almost 70 seconds. Ictal EEG began with bilateral diffuse irregular sharp waves and slow waves, followed by semi-rhythmic bursts of slow waves, which were replaced by recruiting rhythmic waves of alpha range. Left rhythmic waves preceded right-sided waves. These rhythmic waves decreased in frequency at the end. In the late phase, the spikes on the left and right sides did not completely synchronize with each other. A slight lag was observed among spikes from different areas. In the postictal period, the delta waves mixed with artefacts of oral automatism over bilateral temporal regions, continuing with predominance in the left side. Although the ictal EEG looked like the sGTC pattern, surface EMG was missing for both deltoid muscles except in the initial tonic phase.
Discussion
Mutations in PCDH19 have been reported to be associated with various neurological manifestations, which expand the phenotypic variability. We report three patients with convulsive seizure clusters that consisted of a few to dozens of seizures associated with fever. Although hot water immersion-induced seizures have been reported (Higurashi et al., 2011), our patients did not show any such episodes. Moreover, the seizure frequency was not high in our patients, usually occurring every one to three months. The phenotype of PCDH19RE includes features of generalised epilepsy, focal epilepsy, and Dravet syndrome (Scheffer et al., 2008; Depienne et al., 2009, 2011; Marini et al., 2010). PCDH19RE has been reported to include generalised tonic-clonic, focal, tonic, absence, or myoclonic seizures (Higurashi et al., 2011; Specchio et al., 2011; Higurashi et al., 2013). PCDH19RE has also been shown to exhibit a distinctive electroclinical pattern of focal seizures with affective symptoms on ictal video-EEG, however, there are few reported detailed studies of convulsive seizures (Marini et al., 2012). In this report, we investigated convulsive seizures, considered as secondary generalised seizures, that are also seen in PCDH19RE, in addition to focal seizures.
Our detailed analysis of ictal video-EEG recordings revealed a unique sequential manifestation that consists of six phases: “jerk”, “reactive”, “mild tonic”, “fluttering”, “mild clonic”, and “postictal”. Jerks can be seen in the axial body and/or limbs with (Patient 1 and Patient 2) or without (Patient 3) coughing or gurgling sounds. The “jerks” occurred singly or repeated irregularly without spikes or waves on EEG. The second phase is the “reactive” phase, which is like a “panic” or fearful state. The patients sometimes look so startled by the initial sudden jerks during sleep that they try to turn over or sit up. Patient 2 jumped up and tried to fling her arms around a nearby person. This “reactive” or hyperactive phase may represent affective symptoms reported by Marini et al. (2012). Seizure symptoms look like complex gestural automatisms. The third phase is “mild tonic”, which is similar to the tonic phase of the tonic-clonic seizure, but less intense compared with the typical tonic phase because of reduced involvement of the deltoid muscles. It is more or less asymmetric. EEG shows recruiting fast waves originating from unclear or different foci. The “mild tonic” phase is gradually replaced by the fourth phase, “fluttering”, which includes movements which are reminiscent of a baby lying on its back. In this phase, each extremity trembles and jerks in an asymmetric, less rhythmic, and less synchronous manner, with tremulous fingers and hands. In the later phase, the movement of the limbs gradually becomes more synchronous and nearly clonic. The proximal muscle contraction is not as intense as that in typical clonic seizures. In the last phase, “postictal state”, patients become motionless with or without oral automatism. This phase may be associated with postictal bilateral diffuse and continuous slow waves on EEG, especially between seizures in the same clusters. This postictal phase may be misdiagnosed as an atypical absence seizure or prolonged complex partial seizures.
The 19 of the 26 convulsive seizures (4/6: Patient 1; 6/6: Patient 2; 9/14: Patient 3) exhibited all phases. However, convulsive seizures do not necessarily exhibit all of these six phases. Some phases can be shorter or lacking, whereas others can be longer or more pronounced; this may explain the diversity of seizure manifestations reported in the literature. However, these phases do usually occur in sequential order. The most characteristic phases in the convulsive seizures are the “reactive”, “mild tonic”, “fluttering”, and “mild clonic”, predominantly appearing in the distal extremities.
In our study, the convulsive seizures were considered as focal-onset seizures with secondary generalisation. Furthermore, these seizures can originate from either side even in the same patients. We speculate that hyperexcitability of the brain is so widespread and unstable that the seizures easily propagate in PCDH19RE, like the falsely generalised seizures and unstable seizures seen in Dravet syndrome. However, the tonic and clonic phases are less intense and more dominant in the periphery compared with Dravet syndrome. It is noteworthy that most seizures in this study occurred during sleep.
In conclusion, we conducted ictal video-EEG recordings of 26 convulsive seizures in three PCDH19RE patients. Based on our analysis of these recordings, we have found that these seizures consistently progress through six sequential phases. We have also found that the EEG recordings indicate the presence of widespread hyperexcitability throughout the brain in these patients. These characteristic features of the convulsive seizures may help in the diagnosis of PCDH19RE, but a large number of patients may be needed to confirm our findings.
Acknowledgements and disclosures
The authors thank the patients and their families who participated in our study. This study was funded in part by Grants-in-aid for Scientific Research I Nos. 23591238, 24591537 and 26293122, Health and Labour Sciences Research Grants for Comprehensive Research on Disability Health and Welfare (H24-002); Research on rare and intractable diseases; and grants from The Japan Epilepsy Research Foundation. The work of genetic analyses was supported in part by Grant-in-Aids for Scientific Research (A) (#21249062) (SH), Scientific Research (C) (26461552) (NH), Challenging Exploratory Research (# 25670481) (SH), Scientific Research on Innovative Areas (25129708) (SH), and Bilateral Joint Research Projects (SH) from The Japan Society for the Promotion of Science (JSPS). In addition, support included a Grant-in-Aid for Scientific Research on Innovative Areas “Genome) Science” (SH) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); MEXT-supported Program for the Strategic Research Foundation at Private Universities 2013-2017 (NH and SH); Research Grants for Nervous and Mental Disorder (21B-5) (SH), Health and Labour Science Research Grants (21210301 and KB220001) (SH), and a Grant-in-aid for the Research on Measures for Intractable Diseases (No. H22-Nanji-Ippan-49) (TH) from the Ministry of Health, Labour and Welfare; the Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University (SH); Research Grants from Kawano Masanori Memorial Foundation for Promotion of Pediatrics (NH), the Mitsubishi Foundation (SH) and Takeda Scientific Foundation (SH); and Research grants for the Central Research Institute for the Molecular Pathomechanisms of Epilepsy of Fukuoka University and Recommended Projects from Fukuoka University (#117016) (SH).
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. The authors have no conflicts of interests to disclose.
* A portion of this work has been previously presented at the 10th European Congress on Epileptology.