Epileptic Disorders
MENUBenign infantile seizures followed by autistic regression in a boy with 16p11.2 deletion Volume 19, numéro 2, June 2017

Benign infantile seizures (BIS) are a familial or sporadic, infantile-onset epilepsy syndrome, characterized by good response to antiepileptic drugs, spontaneous remission by age 2, normal neurodevelopmental outcome, and, sometimes, association with paroxysmal kinesigenic dyskinesia (PKD) (Vigevano, 2005). A recurrent frameshift heterozygous mutation (c.649dupC, p.Arg217Profs*8) in PRRT2 (locus 16p11.2) underlies most BIS/PKD cases (Lee et al., 2012; Becker et al., 2013). Rarely, sub-microscopic 16p11.2 deletions, a well-established cause of neurodevelopmental disorders that includes cognitive/language delay and autism spectrum disorders (ASD)(Weiss et al., 2008), have also been detected in BIS/PKD (Dale et al., 2011; Weber et al., 2013). Why the same microdeletion results in different phenotypes (i.e. ASD or BIS) remains, however, unclear.
Here, we report a child harbouring a 16p11.2 deletion, presenting with BIS and normal early development, unexpectedly followed by cognitive and autistic regression.
Case study
This 5-year-old boy was the second child of healthy, non-consanguineous parents. Birth and neonatal history were normal. At age 5 months, he displayed a first, afebrile seizure characterized by staring, eye deviation, and bilateral jerks, lasting about one minute and followed by post-ictal sleep. After 24 days, similar seizures recurred in clusters of two episodes per day, and remitted without therapy at age 6.5 months. At that time, the child came to our attention, and displayed normal psychomotor development based on standardized assessment (Bayley Scale of Infant and Toddler Development). Neurometabolic screening and EEG during wakefulness and sleep were normal, and a diagnosis of non-familial BIS was made. At age one, he was able to walk with support and, two months later, first words became evident. We observed a completely normal social and cognitive development up to 18 months, when stereotyped and repetitive behaviours, withdrawal, poor social gaze, and no response to his name became progressively evident, fitting the diagnostic criteria for ASD. Communicative gestures and language gradually disappeared, and a circadian rhythm sleep disorder and behavioural/emotional dysregulation clearly emerged. At age 3, cognitive testing (Griffiths Mental Development Scales) revealed mild-to-moderate intellectual disability (ID), and obvious receptive-expressive language impairments. EEG recordings, metabolic work-up, standard karyotyping, and brain MRI were normal.
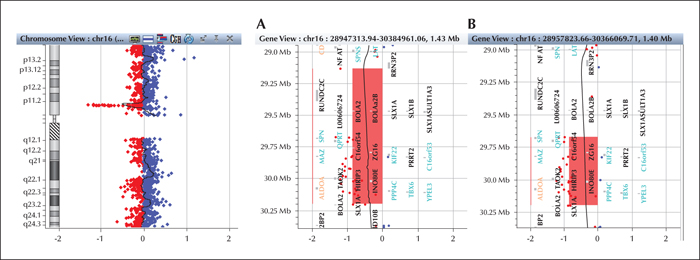
Array-CGH testing, performed after informed consent, detected a 1.064-Mb 16p11.2 deletion (29,133,676-30,198,600) (figure 1A), encompassing about 35 genes. The deletion was inherited from the healthy mother, which differed from her child regarding the first distal oligo mapping in a region of segmental duplications (figure 1B). The mean fluorescent log ratio of the deleted oligos was about -1, consistent with a non-mosaic deletion. Array-CGH in the father showed a 1.2-Mb 19q13.42 duplication (55,913,630-57,188,215).
Discussion
16p11.2 deletions, particularly the recurrent 600-kb deletion defined by breakpoints 4 and 5, havent been associated with up to 1% cases of autism and 1.5% cases of developmental and language delay, thus representing one of the most frequent known syndromic aetiologies for neurodevelopmental disorders and ASD (Weiss et al., 2008). Haploinsufficiency of genes at 16p11.2 may indeed play a role in the development of several brain functions, including language and social cognition (Hanson et al., 2015). However, whilst speech or language-related disorders are extremely common in the syndrome, only 18-25% of deletion carriers fit strict criteria for a diagnosis of ASD (Duyzend and Eichler, 2015; Hanson et al., 2015). Moreover, the degree of ID may be extremely variable, and sometimes carrier individuals may display normal cognitive phenotypes (Hanson et al., 2015). Only rarely, BIS may be the presenting symptom in patients with 16p11.2 deletions covering PRRT2, occurring either with a typical course (Weber et al., 2013) or with a milder seizure history (Dale et al., 2011), similar to our patienthttp://brain.oxfordjournals.org/content/138/12/3476.long - ref-102. Why most PRRT2 heterozygous mutations lead to clinical seizures, while PRRT2 deletion (as in 16p11.2 deletions) does not in most cases is, however, unclear. A dominant negative effect of the mutant PRRT2 allele or the contribution of modifier genes within the deletion are possible explanations for this unsolved question.
Here, we report a child with a 1.064-Mb 16p11.2 deletion, presenting with BIS and normal neurodevelopment followed, at the age of 18 months, by a regression leading to ASD, ID, and language impairment. It is hard to know why, despite the good outcome of seizures, this patient underwent such a severe developmental trajectory. In contrast to single-gene PRRT2 mutations leading to BIS, which do not play a major role in ASD susceptibility (Huguet et al., 2014), the haploinsufficiency of several likely genes included in the deletion may account for a more severe impairment of brain development and function (table 1). However, why the phenotype in this child differed so strongly from that of his asymptomatic mother remains an open issue, possibly due to an increased risk of disease in males with respect to females (Duyzend and Eichler, 2015), inherited versus de novo deletions (Moreno-De-Luca et al., 2015), or incomplete penetrance of 16p11.2 deletions (Rosenfeld et al., 2013). The coding genes in the non-deleted region in the mother (BOLA2, BOLA2B, SLX1B, SLX1A, SULT1A3, and SULT1A4) are not known to be relevant to the phenotype, and are therefore unlikely to contribute to clarifying genotype-phenotype correlations. In addition, the question of whether seizures in our patient may have acted as an epigenetic mechanism which led to gene expression changes, triggering otherwise silent gene imbalances in the deleted region, remains unresolved.
Despite several unresolved issues, this report suggests, however, that the neurodevelopmental prognosis in BIS patients might not be as benign as expected (Becker et al., 2013). It is well established that patients with epilepsy have an increased burden of genomic rearrangements, particularly when associated with ID or other neuropsychiatric problems (Striano et al., 2012). Accordingly, we suggest that infants with BIS, in the absence of mutations in PRRT2, should also be screened for copy number variants in order to rule out 16p11.2 deletions and should be provided with a neurodevelopmental prognosis with caution, with careful clinical follow-up.
Supplementary data.
Summary didactic slides are available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
This study was partially supported by grants from the Italian Ministry of Health (Ricerca Corrente to FS).
None of the authors have any conflict of interest to declare.