Hépato-Gastro & Oncologie Digestive
MENUCancer du pancréas : causes, formes génétiques et dépistage Volume 24, supplément 2, Juin 2017


Université Paris 7, service de gastroentérologie et pancréatologie,
Clichy-La-Garenne, France
- Mots-clés : cancer du pancréas, dépistage, causes, épidémiologie
- DOI : 10.1684/hpg.2017.1445
- Page(s) : 7-17
- Année de parution : 2017
La gravité de l’adénocarcinome pancréatique (AP) du pancréas justifie les efforts faits pour tenter de le diagnostiquer le plus précocement possible, voire de proposer un traitement chirurgical préventif en cas de lésions à risque très élevé de dégénérescence. Il existe trois types de lésions précancéreuses : la néoplasie intra-épithéliale pancréatique (PanIN), la tumeur intracanalaire papillaire et mucineuse du pancréas (TIPMP) et le cystadénome mucineux. L’évolution vers l’AP n’est pas la règle et l’enjeu est doublement important : 1) identifier des signes prédictifs de malignité fiables afin de proposer une chirurgie à bonne escient, et 2) à l’inverse, ne pas opérer une lésion précancéreuse bénigne qui n’évoluera pas, afin d’éviter une chirurgie inutile et délétère.
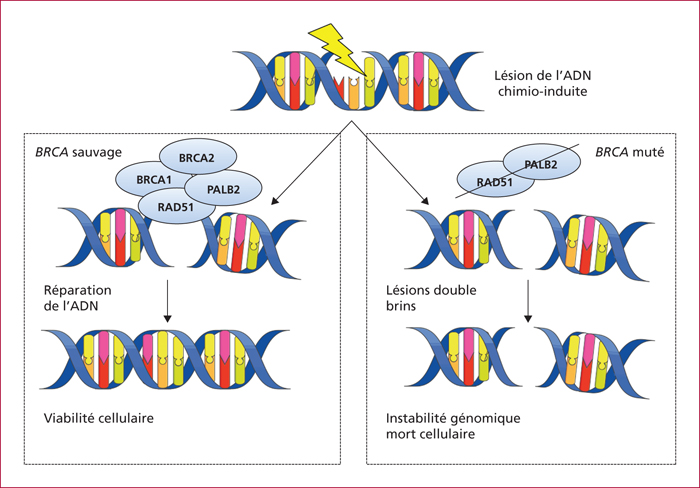
Le risque de développer un AP augmente avec l’âge, dans les pays développés, chez les sujets de sexe masculin et les individus de couleur noire. Les autres facteurs de risque démontrés sont le tabac, l’obésité, le diabète, la pancréatite chronique et les antécédents familiaux d’AP. Les cancers pancréatiques familiaux représentent 5 %-10 % des cas d’AP et sont définis par l’existence d’un AP chez au moins 2 apparentés au premier degré ou de 3 apparentés quel que soit le degré de parenté. Dans ces agrégations familiales, une mutation germinale n’est identifiée que dans environ 15 % des cas, chez qui il existe une forme syndromique avec des mutations germinales de la voie BRCA-Fanconi (gènes BRCA1/2, PALB2, ATM, FANC), ou atteints d’un syndrome de Peutz-Jeghers (gène LKB1/STK11), d’une pancréatite chronique héréditaire (gène PRSS1), d’un mélanome familial multiple (gène p16/CDKN2A) ou d’un syndrome de Lynch. L’identification de ces formes familiales peut justifier un dépistage aux apparentés, une chirurgie pancréatique prophylactique des lésion précancéreuses ou, dans les AP avancés inopérables, une chimiothérapie ou une thérapie ciblée adaptées à la biologie moléculaire de la tumeur, comme un sel de platine ou un inhibiteur de PARP chez les porteurs de mutation de BRCA1/2.