Hépato-Gastro & Oncologie Digestive
MENULa pancréatite auto-immune Volume 24, numéro 8, Octobre 2017

- Mots-clés : pancréatite auto-immmune, IgG4, cholangite slérosante, pancréatite aiguë, adénocarcinome pancréatique, corticothérapie, immunosuppresseurs, rituximab
- DOI : 10.1684/hpg.2017.1505
- Page(s) : 846-56
- Année de parution : 2017
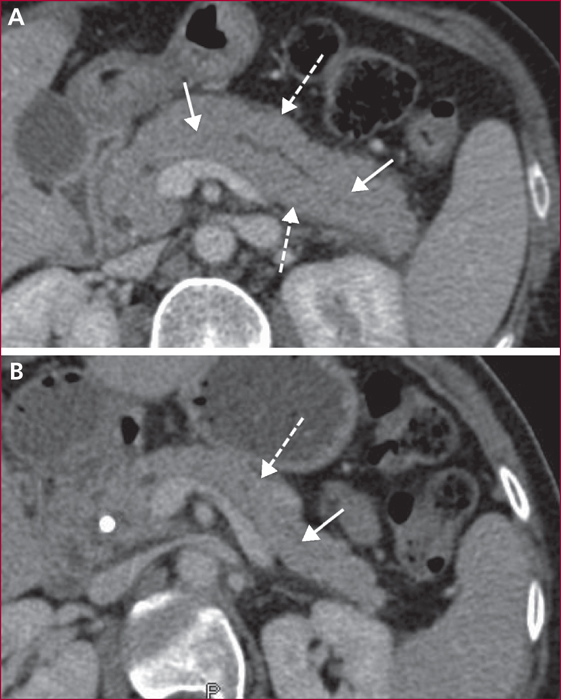
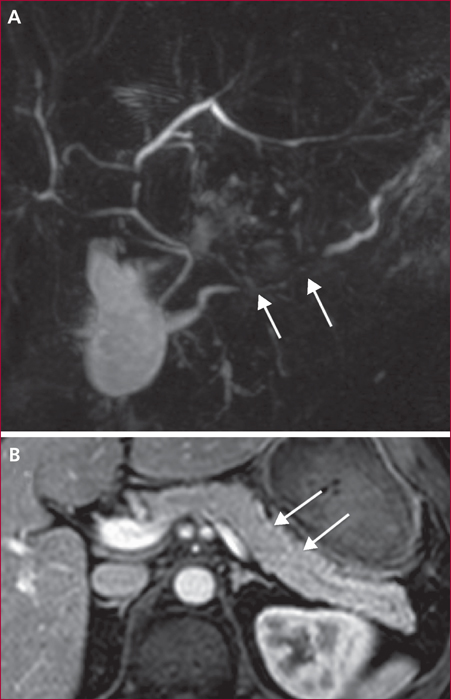
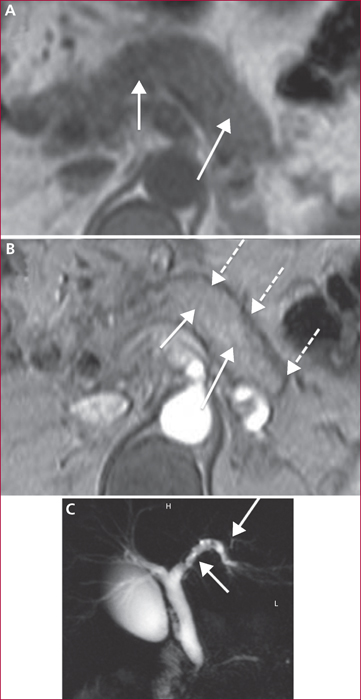
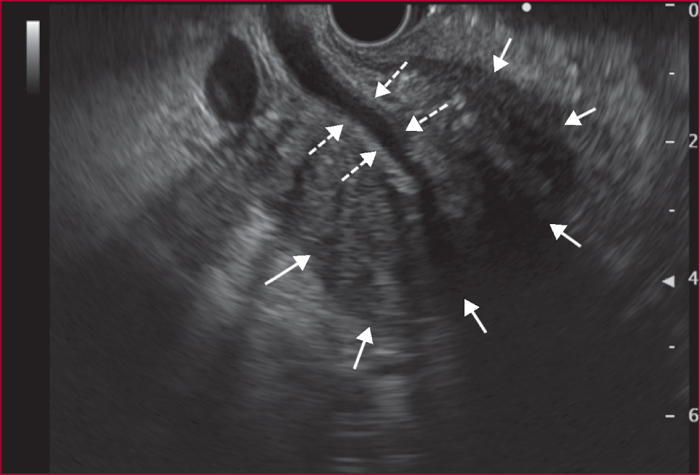
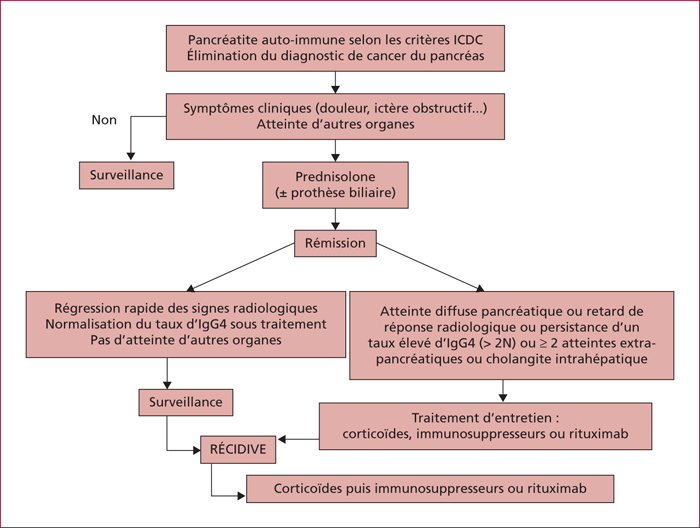
La pancréatite auto-immune (PAI) est divisée en deux entités anatomo-cliniques. La PAI de type 1 associe une atteinte pancréatique, une élévation sérique des IgG4 (≥ 2N) et des atteintes extrapancréatiques (près de 50 % des cas) en particulier les voies biliaires, les glandes salivaire, le rétropéritoine, les reins, les poumons. La PAI de type 2 est une maladie uniquement pancréatique mais une maladie inflammatoire chronique intestinale est associée dans 20 à 30 % des cas. Les signes cliniques des PAI sont un ictère en rapport avec une forme pseudo-tumorale céphalique pancréatique et/ou une cholangite, des douleurs pancréatiques chroniques, des poussées de pancréatites aiguës idiopathiques. Les éléments du diagnostic reposent sur l’imagerie par scanner et IRM avec aspect hypodense/hypo-intense diffus ou localisé avec un halo inflammatoire péri-pancréatique, une quasi-disparition du canal principal ou des sténoses longues ou étagée sans dilatation d’amont. En échoendoscopie, l’atteinte inflammatoire est localisée ou diffuse avec des contours hyperéchogènes et un canal de Wirsung sténosé dont les parois sont épaissies hypo- ou hyperéchogènes. Une cytoponction dirigée peut résoudre parfois les problèmes de diagnostic positif et différentiel avec l’adénocarcinome pancréatique. Le test thérapeutique par prednisolone, durant 12 semaines fait partie du diagnostic avec une régression rapide (2 semaines) et significative des signes cliniques et radiologiques. Si la réponse thérapeutique est obtenue dans 90 % des cas, la récidive est observée dans 10 à 30 % des cas en particulier pour les PAI de type 1, avec manifestations extrapancréatiques (≥ 2, notamment biliaire), un taux élevé d’IgG4 (> 4N) avant traitement. Un nouveau traitement par corticoïdes est alors proposé avec mise en route d’un traitement de fond par immunosuppresseurs (azathioprine ou rituximab). Le pronostic est bon aux prix de séquelles à type d’insuffisances pancréatiques endocrine et exocrine dans environ 30 % des cas.