Annales de Biologie Clinique
MENUDifficulté diagnostique d’un syndrome bêta-thalassémique chez une patiente multi-transfusée : apport du myélogramme et de l’étude des parents Volume 75, numéro 5, Septembre-Octobre 2017

- Mots-clés : bêta-thalassémie, myélogramme, transfusion
- DOI : 10.1684/abc.2017.1284
- Page(s) : 562-8
- Année de parution : 2017
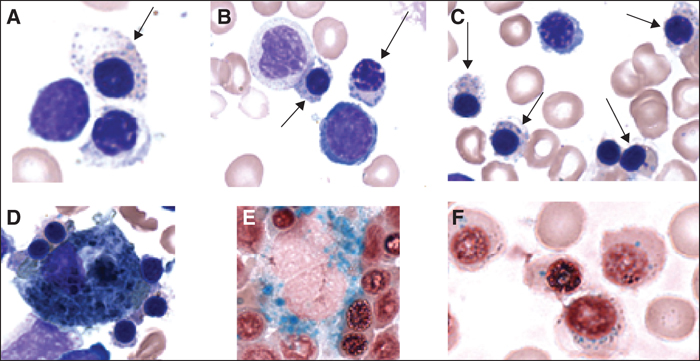
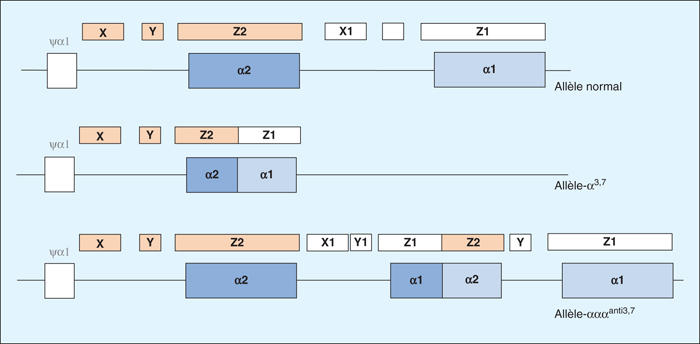
Nous rapportons le cas d’une enfant de 5 ans, initialement suivie pour anémie sidéroblastique congénitale, dont les explorations vont révéler une pathologie familiale de type bêta-thalassémique. Le myélogramme réalisé chez l’enfant met en évidence des érythroblastes matures dystrophiques avec un défaut d’hémoglobinisation et des ponctuations basophiles. Ces anomalies orientent vers une anomalie de synthèse de l’hème ou des chaînes de globines. Les transfusions itératives réalisées chez l’enfant ne permettent pas d’interpréter une recherche d’anomalie de l’hémoglobine. En revanche, la recherche d’anomalie de l’hémoglobine réalisée chez les parents montre une augmentation du taux d’HbA2 associée à une microcytose, concluant à un trait bêta-thalassémique. Sachant que les syndromes bêta-thalassémiques sont des pathologies génétiques, en général récessives, la présence d’un trait bêta-thalassémique chez les parents associée à la clinique de l’enfant (besoin transfusionnel mensuel) sont très en faveur d’un syndrome bêta-thalassémique chez l’enfant. Cette hypothèse diagnostique sera confirmée par l’étude moléculaire des gènes de globine qui va révéler une hémoglobinopathie complexe pour l’ensemble des membres de la fratrie. Les parents sont porteurs à l’état hétérozygote d’une mutation β+thalassémique que l’enfant atteint présente à l’état homozygote permettant de porter le diagnostic de syndrome bêta-thalassémique. De plus, une triplication des gènes α-globine est présente respectivement à l’état hétérozygote chez la mère et à l’état homozygote chez le père et l’enfant. La triplication des gènes α-globine est un facteur connu d’aggravation des bêta-thalassémies et ce cas clinique, avec le continuum clinico-biologique observé, illustre parfaitement les intrications entre gènes α et β globine.