Annales de Biologie Clinique
MENULes bêta-thalassémies : aspects moléculaires, épidémiologiques, diagnostiques et cliniques Volume 72, numéro 6, Novembre-Décembre 2014

Illustrations
-
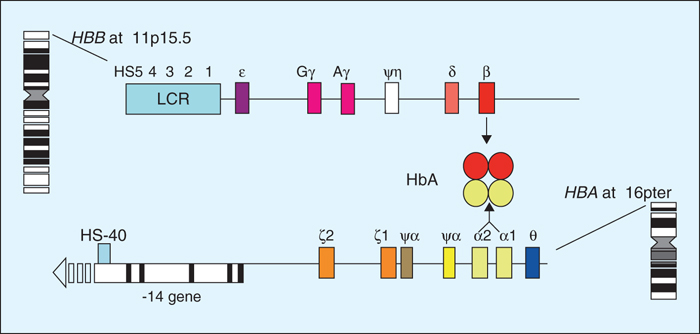
Figure 1 -
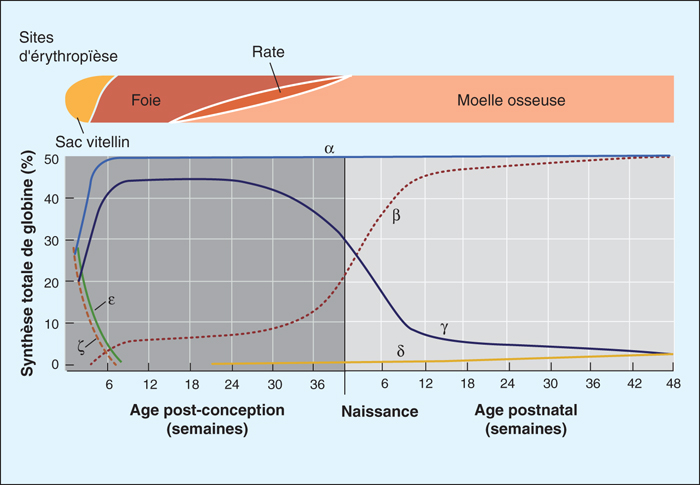
Figure 2 -

Figure 3 -
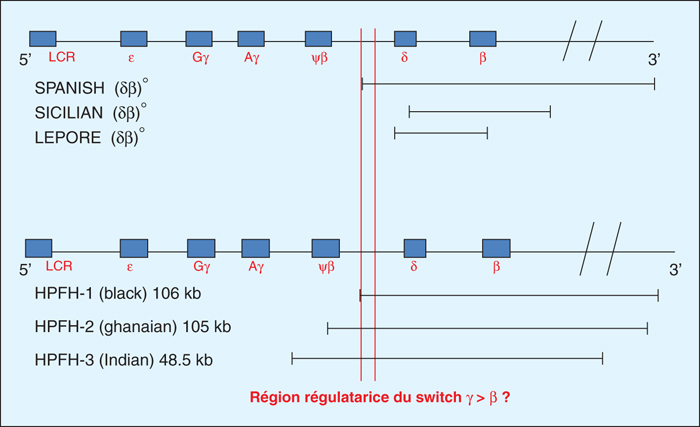
Figure 4 -
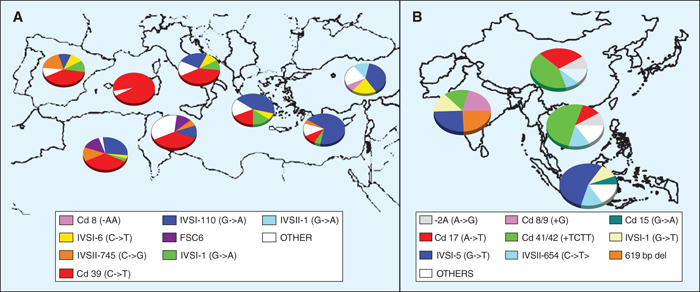
Figure 5 -
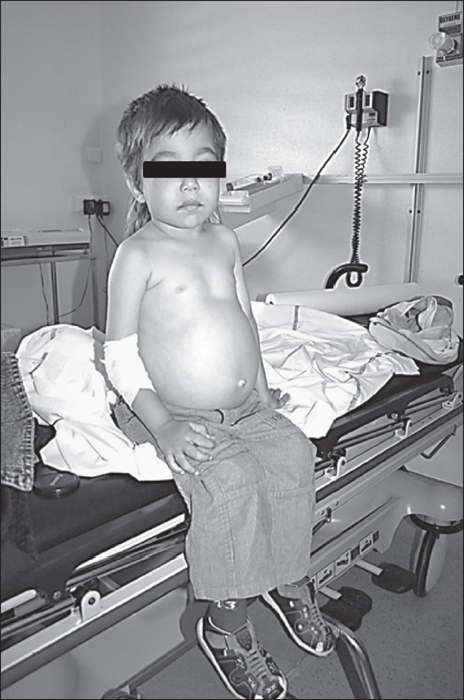
Figure 6 -
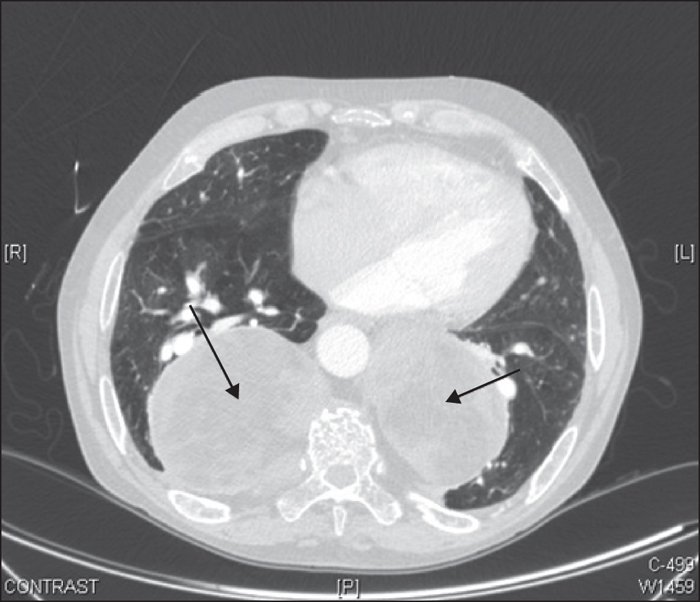
Figure 7 -
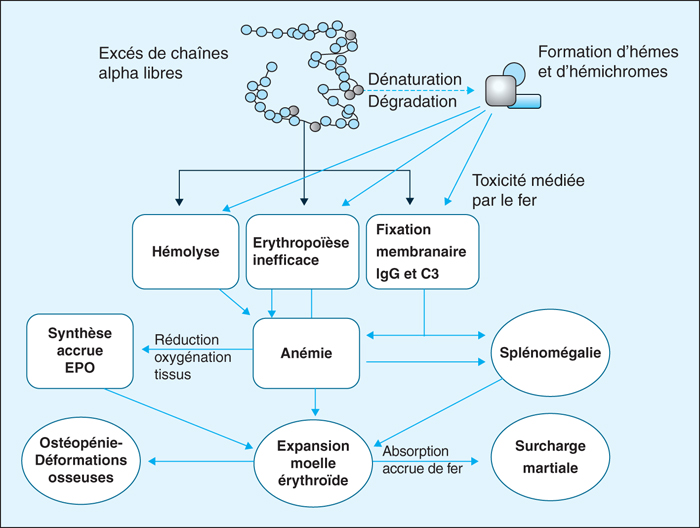
Figure 8 -
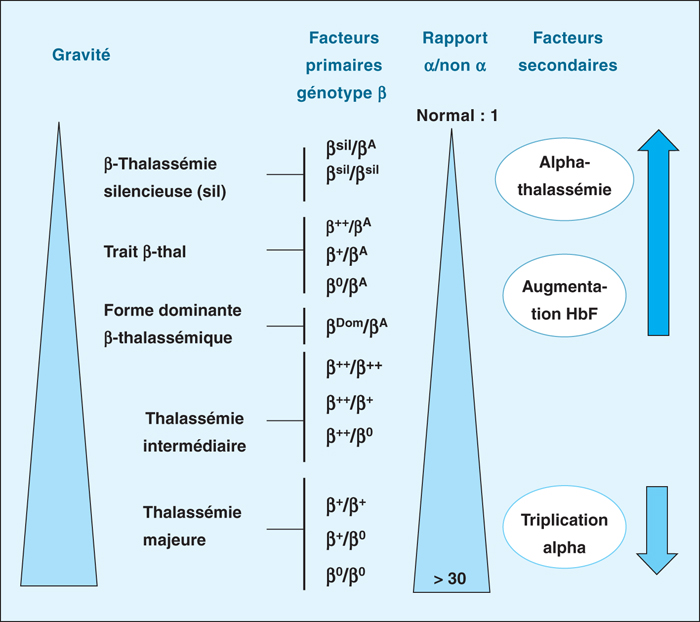
Figure 9 -
Figure 10 -
Figure 11 -

Figure 12 -
Figure 13 -
Figure 14 -

-

-

-

Tableaux
- Mots-clés : bêta-thalassémies, revue, pathologie, diagnostic
- DOI : 10.1684/abc.2014.1015
- Page(s) : 639-68
- Année de parution : 2014
Les bêta-thalassémies représentent une des maladies autosomiques récessives les plus fréquentes dans le monde. En France, on dénombre 5 à 10 nouveaux cas de formes majeures ou intermédiaires par an pour une prévalence globale d’environ 500 malades pour lesquels la généralisation des traitements chélateurs du fer a permis d’augmenter de façon très importante l’espérance de vie depuis une vingtaine d’années. Au niveau moléculaire, environ 90 % des allèles bêta-thalassémiques sont représentés par des mutations ponctuelles caractérisables facilement par séquençage Sanger ou par des techniques dédiées. Les 10 % restants sont des délétions plus ou moins larges détectables par MLPA ou par CGH Array. La détermination du génotype alpha est capitale dans l’exploration génétique d’une bêta-thalassémie puisqu’une alpha-thalassémie améliore la clinique tandis qu’une triplication alpha l’aggrave. Le génotypage additionnel d’autres polymorphismes inducteurs d’HbF permet même, au moyen d’un algorithme dédié, de prévoir l’âge de la première transfusion, faisant de la bêta-thalassémie l’une des premières applications potentielles de la médecine prédictive. Thérapie génique, diagnostic pré-implantatoire et nouveaux traitements médicamenteux (Sotatercept®, agonistes de l’hepcidine) achèvent de placer la β-thalassémie au-devant de l’actualité scientifique.